
Bone marrow neoplastic
Bone marrow - neoplastic myeloid
Myeloid / lymphoid neoplasms with eosinophilia and gene rearrangement
JAK2
Editorial Board Member: Elizabeth Courville, M.D.
Deputy Editor-in-Chief: Genevieve M. Crane, M.D., Ph.D.


Last author update: 8 July 2021
Last staff update: 27 April 2023
Copyright: 2021-2024, PathologyOutlines.com, Inc.
PubMed Search: PCM1-JAK2 fusion
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Peripheral smear description | Peripheral smear images | Molecular / cytogenetics description | Molecular / cytogenetics images | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1Cite this page: Nageshwar M, Singh Z. JAK2. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/bonemarrowneoplasticPCM1JAK2.html. Accessed May 13th, 2024.
Definition / general
- PCM1-JAK2 fusion is a rare genomic abnormality resulting from t(8;9)(p22;p24.1), which fuses the Janus activated kinase 2 gene (JAK2) with the human autoantigen pericentriolar material gene 1 (PCM1), resulting in a constitutively activated tyrosine kinase
- Observed in both myeloid and lymphoid neoplasms and can present as myeloproliferative neoplasm (MPN), myelodysplastic / myeloproliferative neoplasm (MDS / MPN), acute leukemia (B or T lymphoid, myeloid or mixed phenotype), lymphoblastic lymphoma or blast stage of a chronic myeloid neoplasm
Essential features
- Presence of t(8;9)(p22;p24.1) PCM1/JAK2
- Chronic myeloid leukemia and rearrangements of PDGFRA, PDGFRB and FGFR1 must be excluded
- Variable peripheral blood and bone marrow eosinophilia
- Bone marrow morphology; usually like an MPN, MDS / MPN or acute leukemia - both myeloid and lymphoid
- Proliferation of early erythroblasts in the bone marrow
- Dyserythropoiesis may be present
- Marrow reticulin fibrosis
Terminology
- Neoplasms with t(8;9)(p22;p24.1) / PCM1-JAK2 are included as a provisional entity in the 2016 revision of the WHO classification in the category of myeloid / lymphoid neoplasm with eosinophilia and specific gene rearrangements
- Rare cases with other fusion partners of JAK2, such as t(9;12)(p24.1;p13) / ETV6-JAK2 and t(9;22)(p24.1;q11.2) / BCR-JAK2, are considered as variants of this entity
ICD coding
- ICD-O: 9968/3 - Myeloid / lymphoid neoplasms with PCM1-JAK2
Epidemiology
- M:F = 27:5 showing distinct male preponderance
- Wide age range (12 - 75), with median age of 47 years
- Reference: Br J Haematol 2014;166:809
Sites
- Peripheral blood and bone marrow
- Rarely lymph nodes and extramedullary locations
Pathophysiology
- Due to the presentation as both myeloid and lymphoid neoplasms, the cell of origin is purported to be the pluripotential hematopoietic stem cell (Cancer Res 2005;65:2662)
- PCM1-JAK2 fusion mediates an oligomerization that brings together the linked JAK2 domains, resulting in a constitutively active tyrosine kinase domain of JAK2, which activates multiple downstream signal transducers via the JAK-STAT pathway to promote cell proliferation and differentiation
- Underlying pathobiology does not depend on the fusion partner of JAK2
- As far as is known, the translocation partners do not affect the clinical picture, which is determined by the activation of the JAK2 tyrosine kinase
Diagrams / tables
Images hosted on other servers:

Structure of PCM1-JAK2 translocation
Clinical features
- Clinical features are variable depending on the presentation as a chronic myeloid neoplasm, acute leukemia or a lymphoblastic lymphoma (Leukemia 2005;19:1692)
- Common presenting symptoms are fatigue, pallor, weight loss, abdominal discomfort, splenomegaly and lymph node enlargement (Leukemia 2005;19:1692)
- May represent the blastic phase of an underlying chronic neoplasm
- Reference: Ann Lab Med 2016;36:79
Diagnosis
- Review of the CBC and peripheral smear evaluation; if eosinophilia is present, all secondary causes of eosinophilia should be excluded clinically and by appropriate investigation (of note, eosinophilia may not be a prominent feature)
- Flow cytometry to exclude the presence of an immunophenotypically aberrant T cell population as cause of eosinophilia (also known as lymphocyte variant eosinophilia)
- Bone marrow core biopsy and aspirate morphology
- Conventional cytogenetics or FISH to exclude BCR-ABL1 and FISH to detect rearrangements of PDGFRA, PDGFRB and FGFR1
- Identification of the t(8;9)(p22;p24.1) PCM1-JAK2 translocation by conventional cytogenetics and confirmation by FISH or RT-PCR for the fusion gene product
- References: Blood 2017;129:704, Am J Hematol 2019;94:1149
Laboratory
- Peripheral blood evaluation may show anemia or pancytopenia, normal, mildly or markedly increased WBC, leukoerythroblastosis, mild or striking eosinophilia or increased blasts if presentation is as an acute leukemia
- Bone marrow shows myeloid proliferation with maturation; there may be dysplasia in one or more lineages, variable eosinophilia and may show increase in mast cells, usually as loose clusters
- Frequent finding is presence of paratrabecular clusters of early erythroid precursors
- At least some myelofibrosis is frequently present, which is highlighted by the reticulin stain (Eur J Haematol 2011;86:87)
- Metaphase cytogenetics will reveal the characteristic translocation
- In the variant forms of the PCM1-JAK2 translocation involving other fusion partners with JAK2, a JAK2 break apart FISH probe identifying a rearrangement as a first step may be a clue for further FISH studies to identify the specific chromosomal partner
- Fusion is confirmed by PCM1-JAK2 dual color, dual fusion probe or by RT-PCR for the fusion gene product
- In the variant forms of the PCM1-JAK2 translocation involving other fusion partners with JAK2, or in rare cases of crytic rearrangement, a JAK2 break apart FISH probe identifying a rearrangement as a first step may be a clue for further FISH studies to identify the specific chromosomal partner
- Next generation sequencing usually does not reveal additional somatic mutations (Am J Clin Pathol 2021;155:160)
Prognostic factors
- Aggressive behavior; usually does not respond to imatinib
- Good response has been seen with JAK1/2 inhibitor ruxolinitib, although eventually allo-SCT is required
- Prognosis is quite variable (from few weeks to > 5 years) or longer if patient undergoes allogenic bone marrow transplant
- Reference: Blood 2012;120:1529
Case reports
- 35 year old man presented with a history of abdominal discomfort, early satiety, low grade fever, night sweats and weight loss (Blood 2013;122:861)
- 42 year old man with left inguinal and multiple cervical lymphadenopathy; a CT scan identified splenomegaly (Ann Lab Med 2016;36:79)
- 51 year old woman was diagnosed as having acute myeloid leukemia at a tertiary hospital (Ann Lab Med 2018;38:492)
- 57 year old man presented with fatigue, weight loss and dyspnea for 3 months; clinical examination only revealed a splenomegaly (Eur J Haematol 2011;86:87)
Treatment
- Neoplasms resulting from PCM1-JAK2 mutation do not respond to imatinib; the JAK1/2 inhibitor ruxolitinib has shown good response, although eventually these patients require allogenic stem cell transplantation
Microscopic (histologic) description
- Classic triad features of bone marrow biopsy are: hypercellularity with eosinophilic infilterate, aggregates of immature erythroblasts and frequent marrow fibrosis; this triad may not be seen in variants with fusion of ETV6-JAK2 and BCR-JAK2 (Am J Clin Pathol 2021;155:160)
- Variable morphology: may be of a chronic myeloid neoplasm or of acute leukemia; the latter may develop as a blast crisis of the chronic myeloid neoplasm
- Some cases may not have eosinophilia
- Megakaryocytes may be increased or decreased but are often dysplastic
- Variable grades of reticulin fibrosis; usually present
- Reference: Eur J Haematol 2011;86:87
Microscopic (histologic) images
Contributed by Zeba N. Singh, M.B.B.S., M.D.

Markedly hypercellular bone marrow

Megakaryocytic atypia
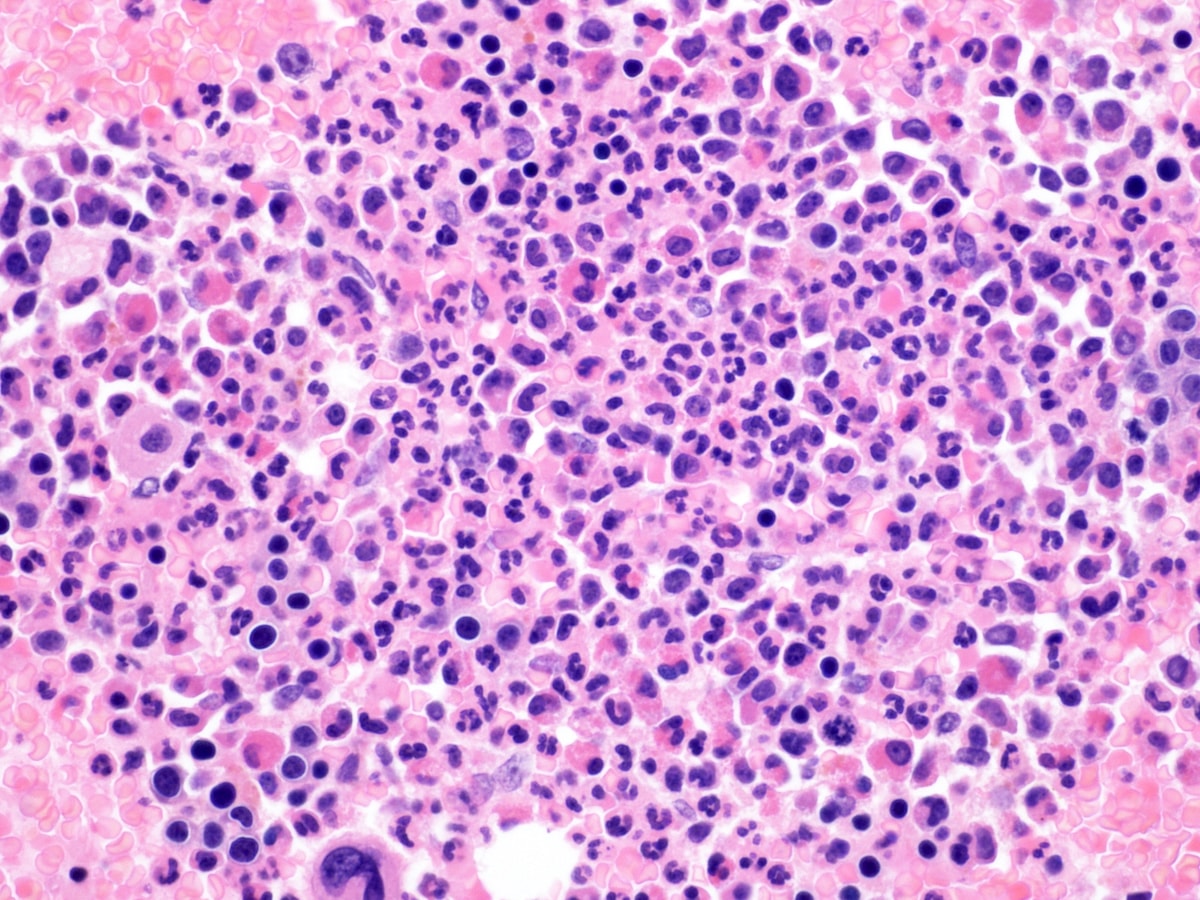
Bone marrow eosinophilia
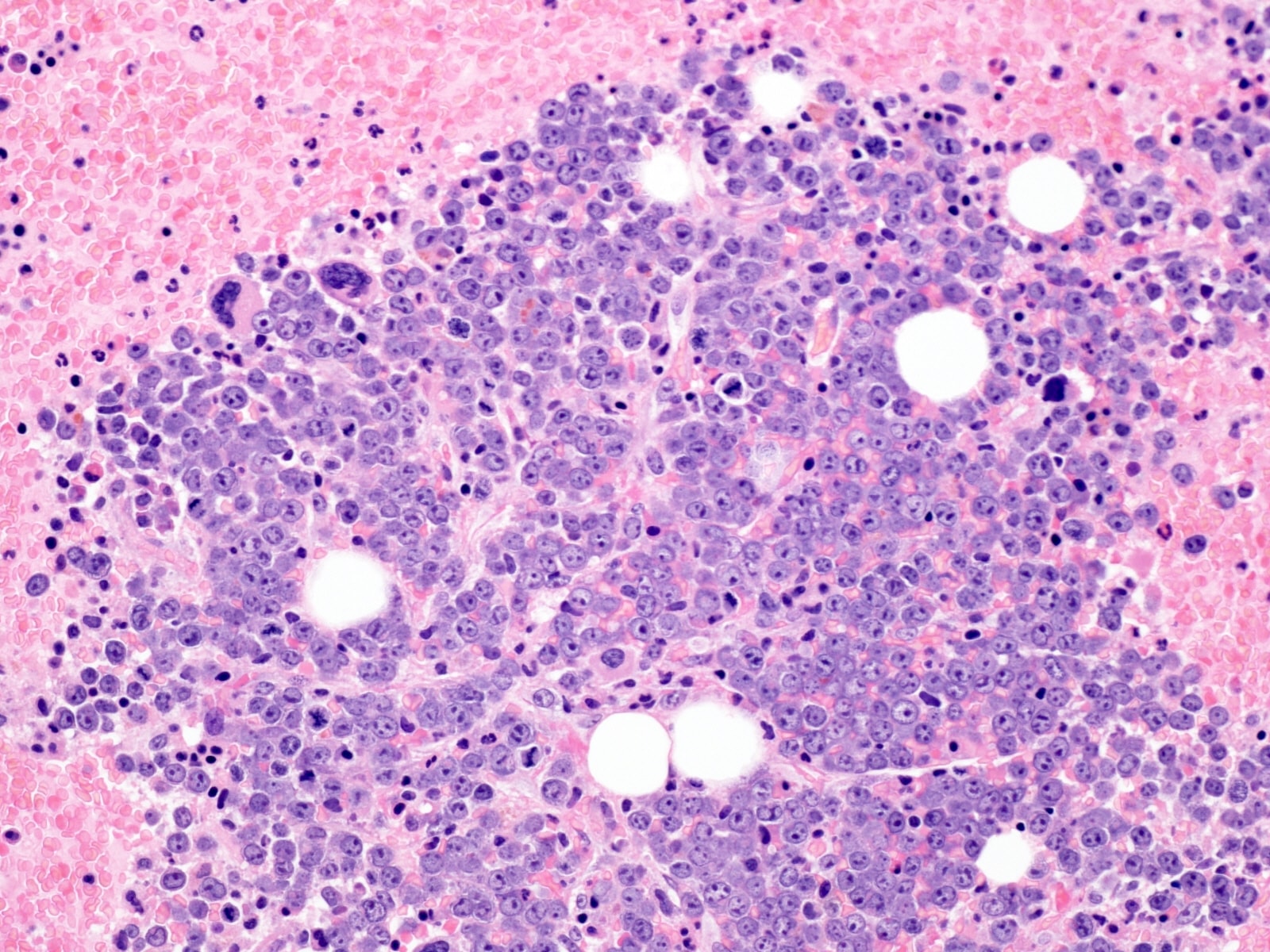
Large aggregate of early erythroid precursors
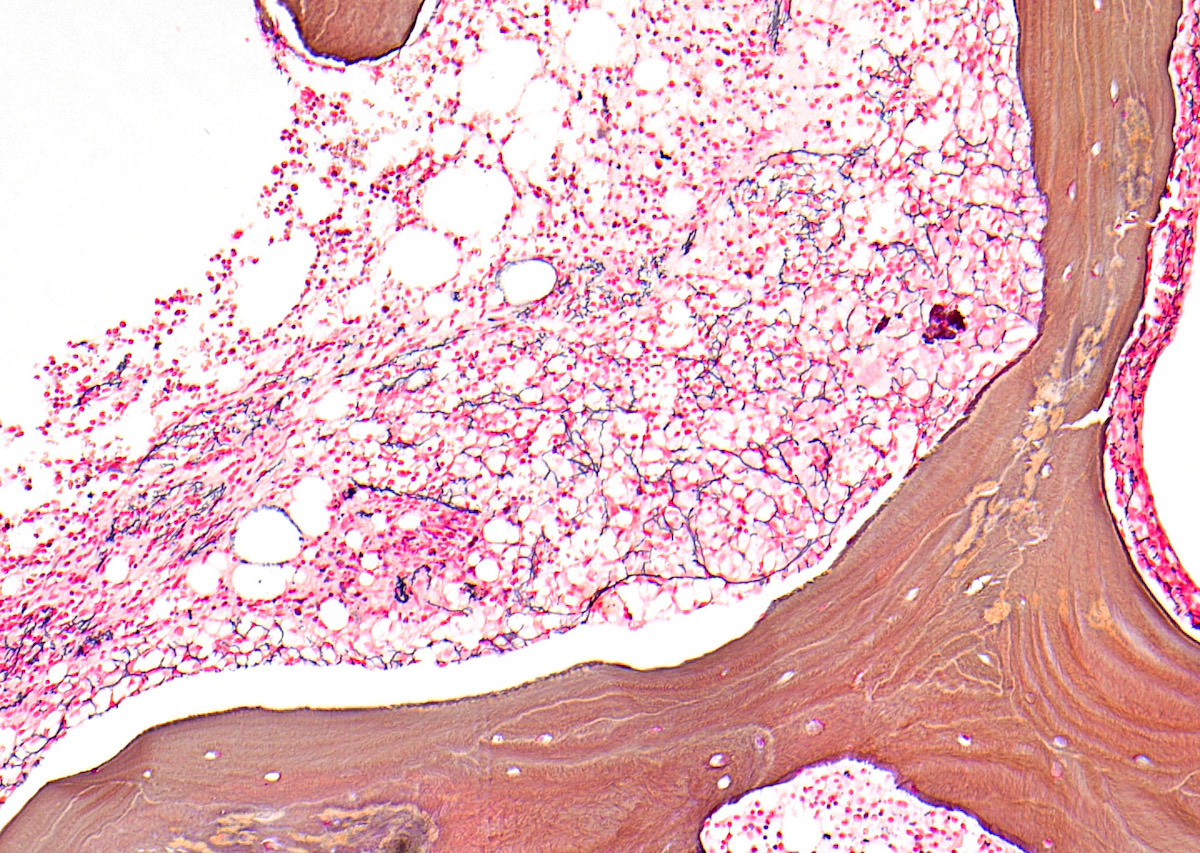
Reticulin fibrosis

CD34 immunostain
Peripheral smear description
- Normochromic, normocytic anemia
- Leukocytosis, leukopenia or leukoerythroblastic picture
- Eosinophilia often but not always
- In cases that present as acute leukemia, circulating blasts are present
- Reference: Am J Hematol 2019;94:1149
Peripheral smear images
Contributed by Zeba N. Singh, M.B.B.S., M.D.
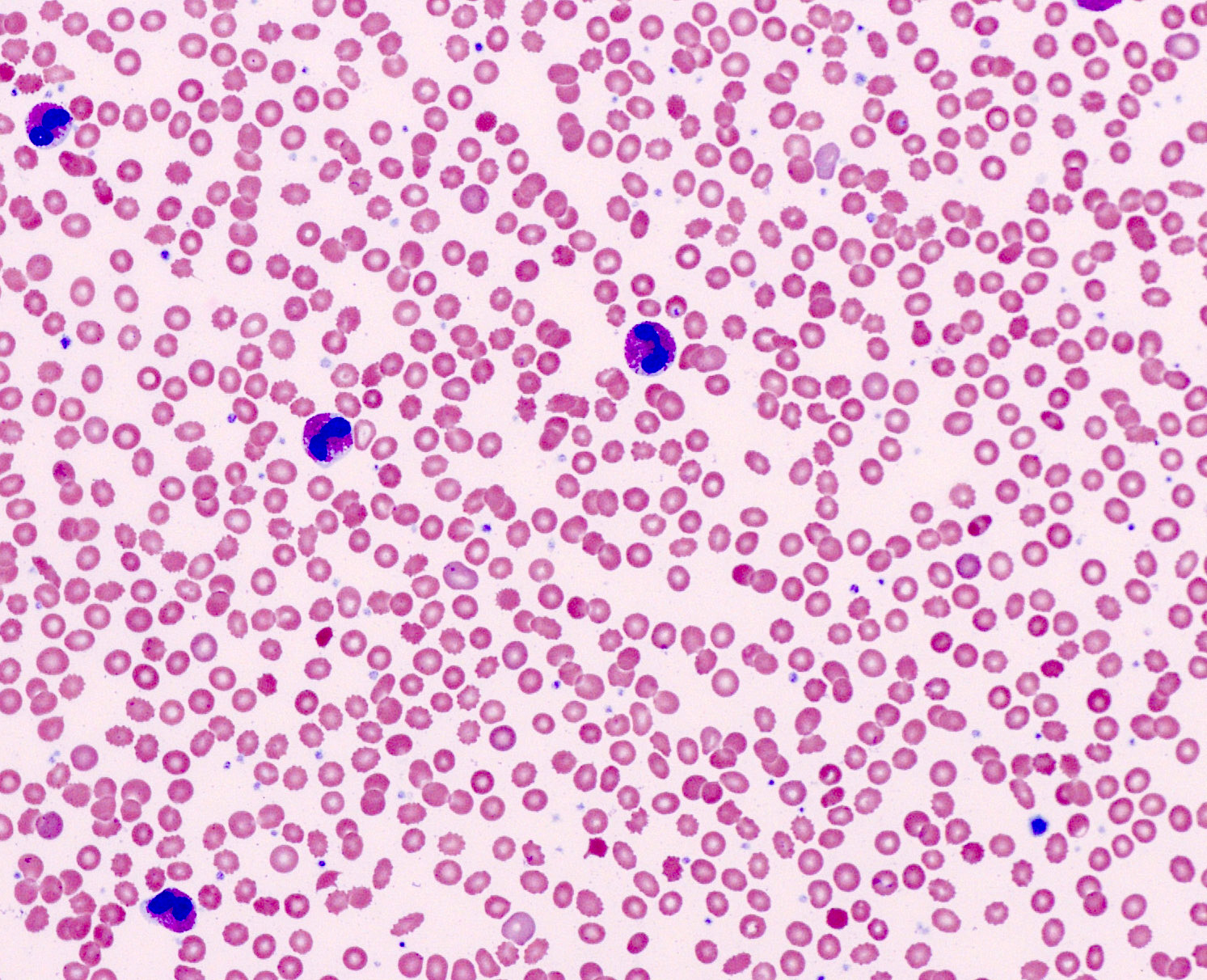
Peripheral blood eosinophilia
Molecular / cytogenetics description
- PCM1-JAK2 fusion mediates an oligomerization that brings together the linked JAK2 domains resulting in a constitutively active tyrosine kinase
- JAK2 is a cytoplasmic tyrosine kinase; it activates multiple downstream signal transducers via the JAK-STAT pathway to promote cell proliferation and differentiation
- BCR-JAK2 fusion protein contains the coiled coil dimerization domain of BCR and the protein tyrosine kinase domain (JH1) of JAK2
- t(8;9)(p22;p24.1) resulting in PCM1-JAK2 fusion
- t(9;12)(p24.1;p13.2) resulting in ETV6-JAK2
- t(9;22)(p24.1;q11.2) resulting in fusion of BCR-JAK2
- Reference: Cancer Res 2005;65:2662
Molecular / cytogenetics images
Images hosted on other servers:
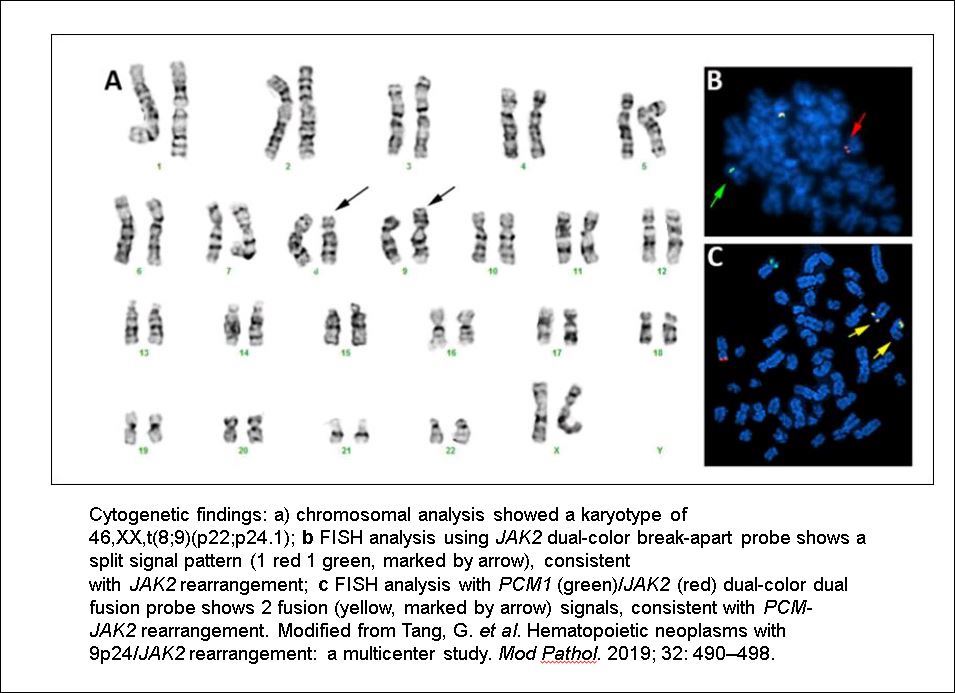
Karyogram and FISH analysis
Sample pathology report
- Bone marrow, right posterior iliac crest, core biopsy and clot section:
- Chronic myeloid neoplasm with t(8;9)(p22;p24.1) PCM1-JAK2 fusion (see comment)
- Hypercellular (90%) bone marrow showing myeloid hyperplasia, dysmegakaryopoiesis, eosinophilia and reticulin fibrosis (grade 1/3); blasts less than 5%.
- Comment: The morphological features of the bone marrow core biopsy and clot section are consistent with a myelodysplastic / myeloproliferative neoplasm, unclassifiable (MDS / MPN-U). There is peripheral blood eosinophilia and an increase in marrow eosinophils. Cytogenetic analysis identified t(8;9)(p22;p24.1) abnormality in 20 / 20 metaphases. RT-PCR for the chimeric BCR-ABL1; FISH for PDGFR alpha, PDGFR beta, FGFR1 rearrangements and JAK2 V617F mutation were negative. The patient had hepatosplenomegaly, pulmonary, cardiac and bone involvement.
- The t(8;9)(p22;p24.1) abnormality fuses the PCM1 gene with the JAK2 gene. Based on the WHO classification (2016 revision), the neoplasm is classified under the group of myeloid / lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, FGFR1 rearrangements or t(8;9) PCM1-JAK2 fusion. The neoplasm is aggressive in behavior. Response to ruxolitinib has been demonstrated (Blood 2012;120:1529).
- Peripheral smear: Manual review of the peripheral blood shows normochromic, normocytic anemia, thrombocytopenia, relative (11%) and absolute (1.65 K/uL) eosinophilia. RBCs: Mild normochromic, normocytic anemia with minimal anisopoikilocytosis. WBCs: There is mild leukocytosis with absolute eosinophilia. The granulocytes show normal morphology. There is a mild left shift with few metamyelocytes and myelocytes. Blasts are not present. The lymphocytes are mature. There is no increase in large granular lymphocytes. Platelets are mildly reduced (100 K/uL) and include some large forms.
- Bone marrow biopsy: Quality: adequate. Cellularity: 90%. Hematopoiesis: full spectrum trilineage hematopoiesis. There is myeloid hyperplasia. There is a prominence of eosinophils and their precursors. Large, paratrabecular aggregates of early erythroid precursors are identified. There is no overt increase in blasts. Megakaryocytes are normal in number but have dysplastic morphology including nuclear hypolobation, hyperchromasia, loose clustering and paratrabecular localization. There is no increase in lymphocytes or plasma cells. Special stains: reticulin; loose network of reticulin without significant intersections (grade 1 fibrosis on a scale of 0 - 3). Trichrome: negative for collagen deposition.
- Bone marrow clot section: Quality: adequate. Cellularity: 90% morphologic features are similar to the core biopsy. Bone marrow aspirate: Quality: adequate. Spicules present. Myeloid hyperplasia. M:E ratio increased (5:1). Full maturation in myeloid and erythroid series. No dysgranulopoiesis or increase in blasts. Eosinophils and eosinophil precursors are increased. Clusters of early erythroid precursors present. Dysmegakaryopoiesis with hypolobated nuclei.
- Immunohistochemistry: Immunohistochemical stains were performed on paraffin fixed and decalcified bone core biopsy sections using appropriate controls. CD34 marks mostly vascular endothelium. Occasional blasts (less than 5%) are identified. CD117 marks the blasts, promyelocytes, early erythroid precursors and mast cells (15%). E-cadherin marks the large aggregates of early erythroid precursors.
Differential diagnosis
- Reactive eosinophilia:
- Nonclonal, secondary cause for eosinophilia (e.g. allergic conditions, parasitic infections, drugs, etc.)
- Detailed clinical history should be taken for exclusion of an underlying cause of eosinophilia
- Chronic myeloid leukemia:
- Uncommonly CML may have absolute eosinophilia
- Absence of BCR-ABL1 fusion excludes CML
- Chronic myelomonocytic leukemia (CMML):
- Absolute monocytosis of 1000/uL or more and absence of genetic abnormalities defining CM, or neoplasms with rearrangements of PDGFRA, PDGFRB and FGFR1; both of these entities are excluded by absence of specific genetic abnormality
- Myeloid neoplasms with PDGFRA, PDGFRB or FGFR1:
- These neoplasms share many morphological features but are distinguished by the specific genetic abnormality
- Chronic eosinophilic leukemia, NOS:
- Persistent eosinophilia, clonal in etiology but not one of the chromosomal translocations included in the entities listed above
- Systemic mastocytosis:
- May be associated with peripheral blood, bone marrow and tissue eosinophilia; bone marrow shows more multifocal aggregates of mast cells with abnormal immunophenotype (CD25+, CD2 variable)
- May be associated with non mast cell clonal hematological disorders
- Lack the specific abnormalities (PDGFRA, PDGFRB, FGFR1 rearrangements or PCM1-JAK2 translocation)
- Idiopathic hypereosinophilic syndrome:
- Peripheral blood eosinophil ≥ 1,500/uL associated with tissue damage, after exclusion of secondary causes of eosinophilia and morphological, flow cytometric, cytogenetic and molecular evaluation for clonal evidence for an acute or chronic myeloid or lymphoproliferative disorder
- Other entities to consider: atypical chronic myeloid leukemia, T and B acute lymphoblastic leukemia and acute myeloid leukemia; all of these entities can be excluded by specific genetic abnormality
- Lymphocyte variant eosinophilia:
- Flow cytometry shows aberrant T cell population
Board review style question #1
A 60 year old man with leukocytosis, mild thrombocytopenia and a differential leukocyte count (showing neutrophils 55%, bands 4%, lymphocytes 22%, monocytes 7%, eosinophils 12%, basophils 1%, metamyelocytes 2%, myelocytes 2% and blasts 1%) undergoes a bone marrow evaluation. Images from the bone marrow core biopsy are provided. Which set of investigations is the most appropriate?
- FISH for inv(16)(13.1;q22), 11q23 / MLL and t(8;21)
- Metaphase cytogenetics, comparative genomic hybridization and next generation sequencing for myeloid mutations
- Metaphase cytogenetics, FISH for PDGFRA, PDGFRB and FGFR1 rearrangement
- qRT-PCR for BCR-ABL1, FISH for NPM1-ALK, FLT3 ITD
Board review style answer #1
C. Metaphase cytogenetics, FISH for PDGFRA, PDGFRB and FGFR1 rearrangement
Comment Here
Reference: PCM1-JAK2
Comment Here
Reference: PCM1-JAK2
