
Bone marrow nonneoplastic
Benign changes
Autoimmune myelofibrosis (AIMF)
Editorial Board Member: Anamarija M. Perry, M.D.


Last staff update: 4 April 2024 (update in progress)
Copyright: 2023-2024, PathologyOutlines.com, Inc.
PubMed Search: Autoimmune myelofibrosis
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Peripheral smear description | Peripheral smear images | Positive stains | Flow cytometry description | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Parunyan M, Vergara-Lluri M. Autoimmune myelofibrosis (AIMF). PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/bonemarrownonneoplasticAIMF.html. Accessed May 2nd, 2024.
Definition / general
- Primary autoimmune myelofibrosis (pAIMF): nonneoplastic myelofibrosis with peripheral blood cytopenias and positive autoimmune antibody serologies, typically in the absence of marked splenomegaly, teardrop poikilocytosis, leukoerythroblastosis, cytologic atypia and a defined autoimmune disorder (Am J Hematol 2003;72:8, Hum Pathol 2014;45:2183)
- Secondary autoimmune myelofibrosis (sAIMF): same features of pAIMF but in the setting of a defined autoimmune disorder (Acta Haematol 2017;138:129)
Essential features
- Nonneoplastic diffuse marrow fibrosis with positive antibody serologies requiring close clinicopathologic correlation for definitive diagnosis
- Usual morphologic features: cytopenia(s) with hypercellular marrow, megakaryocytic and / or erythroid hyperplasias, benign lymphoid aggregates, polytypic plasmacytosis, no or mild hematopoietic dysplasia, mild reticulin fibrosis (MF grade 1)
- No evidence of clonality on ancillary studies (by flow cytometry, chromosomal analysis or next generation sequencing panels for somatic myeloid mutations)
- Usually no or mild splenomegaly
- Typical cases are responsive to glucocorticoids and other immunosuppressive therapy
Terminology
- Systemic lupus erythematosus (SLE) associated myelofibrosis (sAIMF) (J Clin Pathol 1983;36:1219, Medicine (Baltimore) 1994;73:145, Br J Haematol 2007;139:351)
ICD coding
- ICD-10: D75.81 - myelofibrosis
Epidemiology
- Rare
- Incidence and prevalence unknown
- 139 adult cases identified in literature review (Eur J Haematol 2023;111:706)
- Possibly under recognized (Acta Haematol 2017;138:129)
- At least 20 pediatric cases in the literature (Pediatr Blood Cancer 2023;70:e30144, J Clin Rheumatol 2021;27:S378, EJHaem 2020;1:304)
- F:M = ~4:1 (Eur J Haematol 2023;111:706)
- Any age (range: < 1 - 78 years)
- Adult cases (median): 40 - 42 years (Eur J Haematol 2023;111:706)
- Pediatric cases (median): 4.9 - 16.5 years (Pediatr Blood Cancer 2023;70:e30144, J Clin Rheumatol 2021;27:S378)
- ~76% associated with autoimmune syndromes (i.e., sAIMF) (Eur J Haematol 2023;111:706)
Sites
- Bone marrow
Pathophysiology
- Specific pathophysiologic mechanism is not well understood
- Few studies have addressed this question; hypotheses include
- Fibrosis likely driven by overproduction of cytokines
- Predominant source of cytokines is hypothesized to relate to lymphoid aggregates (Clin Adv Hematol Oncol 2018;16:619)
- CD8+ T lymphocytes and IFN gamma have been implicated as having pathogenic roles in a mouse model of primary biliary cholangitis with myelofibrosis (J Autoimmun 2018:89:101)
- IFN gamma signaling through JAK-STAT pathway may play a role
- Overactivation of the JAK-STAT pathway has been associated with multiple autoimmune disorders and malignancies (J Allergy Clin Immunol 2021;147:814)
- Case reports of refractory pAIMF that were responsive to JAK inhibitors (Am J Hematol 2021;96:E283)
- Most hypotheses are extrapolated from studies of other neoplastic myelofibrotic disorders
- Megakaryocytes, platelets and monocytes have been shown to play a role in other etiologies of myelofibrosis (e.g., thrombopoietin associated myelofibrosis, hairy cell leukemia, primary myelofibrosis) via cytokines possibly including PDGF, calmodulin, TGF beta, VEGF and basic fibroblast growth factor (Br J Haematol 2007;139:351, N Engl J Med 2000;342:1255)
- Specific roles of these cytokines in AIMF have not yet been shown
- Increased levels of IFN gamma, TGF beta and IL8 have been associated with cases of primary myelofibrosis (PMF) with autoimmune features (e.g., cases of PMF with positive mitogen stimulated DAT or serologic markers of autoimmunity)
- Unclear if this represents mechanistic overlap between the 2 entities (Leuk Res 2013;37:1509)
- Megakaryocytes, platelets and monocytes have been shown to play a role in other etiologies of myelofibrosis (e.g., thrombopoietin associated myelofibrosis, hairy cell leukemia, primary myelofibrosis) via cytokines possibly including PDGF, calmodulin, TGF beta, VEGF and basic fibroblast growth factor (Br J Haematol 2007;139:351, N Engl J Med 2000;342:1255)
- Fibrosis likely driven by overproduction of cytokines
Etiology
- Unknown
- Role of potential driver mutations is unknown (Haematologica 2021;106:871)
Clinical features
- Symptomatic
- Anemia (83%)
- Thrombocytopenia (34%)
- Fever (30%)
- Signs / symptoms of autoimmune flare
- Raynaud phenomenon, joint pain, peripheral edema
- Night sweats (6%)
- Rarely splenomegaly, usually mild (1%)
- Asymptomatic (at least 32% of patients)
- sAIMF: may present with clinical features of other autoimmune syndromes (Hum Pathol 2014;45:2183, Eur J Haematol 2023;111:706, Haematologica 2021;106:871)
- Most commonly with systemic lupus erythematosus (SLE)
- Also described in patients with acute inflammatory demyelinating polyneuropathy (AIDP), antiphospholipid syndrome (APLS), autoimmune demyelinating polymyositis, autoimmune hemolytic anemia (AIHA), autoimmune hepatitis, dermatomyositis, diabetes mellitus type I (T1DM), Evans syndrome, Hashimoto thyroiditis, immune thrombocytopenic purpura (ITP), mixed connective tissue disease (MCTD), multiple sclerosis (MS), polymyositis, primary sclerosing cholangitis (PSC), psoriasis, rheumatoid arthritis (RA), Sjögren syndrome (SS), vasculitis, vitiligo
- pAIMF: no established autoimmune disorder but all other features associated with sAIMF (Am J Hematol 2003;72:8)
Diagnosis
- Bone marrow biopsy
- May be a dry tap (Clin Adv Hematol Oncol 2018;16:619)
- Peripheral blood smear
- Serologic studies (e.g., autoantibodies)
- Imaging studies (e.g., evaluating spleen)
- Flow cytometry analysis
- Molecular testing (e.g., absent clonal markers of other disorders) (Haematologica 2021;106:871)
- Clinical correlation
Laboratory
- Peripheral cytopenias
- Anemia, thrombocytopenia and / or leukopenia (Eur J Haematol 2023;111:706)
- Serologic autoantibodies
- Cases described with positive antinuclear antibody (ANA), cardiolipin antibodies, cyclic citrullinated peptide antibodies (CCP), direct antiglobulin test (DAT), double stranded DNA antibody (dsDNA), lupus anticoagulant, myeloperoxidase antibodies (MPO), atypical perinuclear antineutrophil cytoplasmic antibody patterns (pANCA), rheumatoid factor (RF), Sjögren syndrome antibodies (SSA / SSB) and / or smooth muscle antibody (Acta Haematol 2017;138:129)
- Elevated inflammatory markers often reported
- E.g., erythrocyte sedimentation rate (ESR) and ferritin (Eur J Haematol 2023;111:706)
- Other abnormal serologic markers
- Hypocomplementemia (Haematologica 2021;106:871)
Prognostic factors
- Overall good prognosis (Acta Haematol 2017;138:129)
- ~85% of cases with clinical follow up had at least partial symptomatic improvement after treatment (Eur J Haematol 2023;111:706)
- Few reports of long term follow up available (Acta Haematol 2017;138:129, Eur J Haematol 2023;111:706)
- Cases of relapse have been reported (Haematologica 2021;106:871, Pediatr Blood Cancer 2022;69:e29762)
- Factors associated with treatment resistance are unknown
Case reports
- 15 year old boy with headaches and severe pallor (EJHaem 2020;1:304)
- 44 year old woman with fatigue, joint pains, oral ulcers, sicca syndrome and malar rash (BMJ Case Rep 2019;12:bcr-2018-227520)
- 48 year old woman with anemia and fever (Am J Clin Pathol 2001;116:211)
- 61 year old woman with severe fatigue (Am J Hematol 2021;96:E283)
Treatment
- Insufficient data on optimal indications and efficacy of different treatment regimens (Haematologica 2021;106:871)
- Adequate prospective trials are difficult due to rarity of AIMF (Eur J Haematol 2023;111:706)
- Usually responds to glucocorticoids
- Most cases with post treatment biopsy data report diminished or reversed marrow fibrosis (Eur J Haematol 2023;111:706)
- Other immunomodulators are sometimes used, alone or with glucocorticoids
- E.g., hydroxychloroquine, mycophenolate, intravenous immunoglobulin, rituximab and others (Eur J Haematol 2023;111:706)
- Ruxolitinib reportedly used in few case reports (Leuk Lymphoma 2023;64:1723, Am J Hematol 2021;96:E283)
- Treatment may aim to control underlying autoimmune disorder (Pediatr Blood Cancer 2023;70:e30144)
Microscopic (histologic) description
- Constellation of morphologic findings may suggest the diagnosis in appropriate clinical context
- Usually mild to moderate reticulin fibrosis of bone marrow
- Mild / delicate fibrosis (MF1; 86%), moderate fibrosis (MF2; 10%), marked fibrosis (MF3; 4%) (Hum Pathol 2014;45:2183)
- Using European consensus classification on grading bone marrow fibrosis (Haematologica 2005;90:1128)
- Mild / delicate fibrosis (MF1; 86%), moderate fibrosis (MF2; 10%), marked fibrosis (MF3; 4%) (Hum Pathol 2014;45:2183)
- Usually hypercellular (72%) or normocellular (24%) bone marrow (Hum Pathol 2014;45:2183)
- Usually with erythroid and megakaryocytic hyperplasia (76% each)
- May show granulocytic hyperplasia (34%)
- Benign lymphoid aggregates (83%) or interstitial infiltrate of lymphocytes (17%) usually present (either / or: 100%; n=29) (Hum Pathol 2014;45:2183)
- Lymphoid aggregates
- Usually small to medium sized
- Nonparatrabecular location
- Well circumscribed
- T and B cell distribution usually in T cell pattern (patterns 1 - 3) (Hum Pathol 2014;45:2183, Hum Pathol 2013;44:512)
- Lymphoid aggregates
- Mild polyclonal / polytypic plasmacytosis may be present (66%), typically < 10% plasma cells (Hum Pathol 2014;45:2183, Am J Hematol 2003;72:8)
- No significant osteosclerosis or bony changes
- No or minimal dysplasia of hematopoietic lineages
- Occasional atypical forms may be present
- Should not meet diagnostic criteria for myeloid neoplasms
- No monoclonal / monotypic population
- No increase in blasts
- Reference: Hum Pathol 2014;45:2183
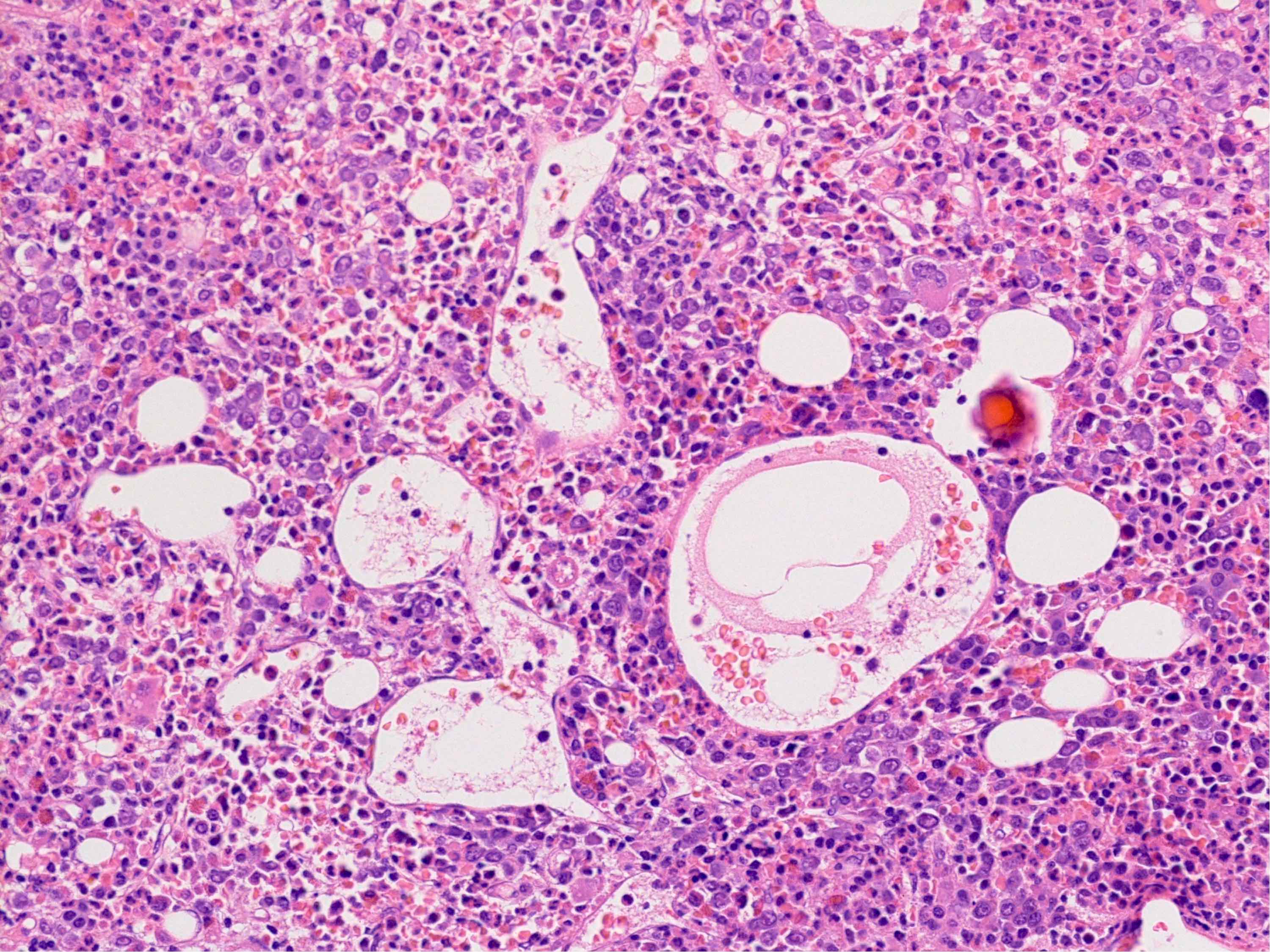
Microscopic (histologic) images
Contributed by Ria Vergara-Lluri, M.D.
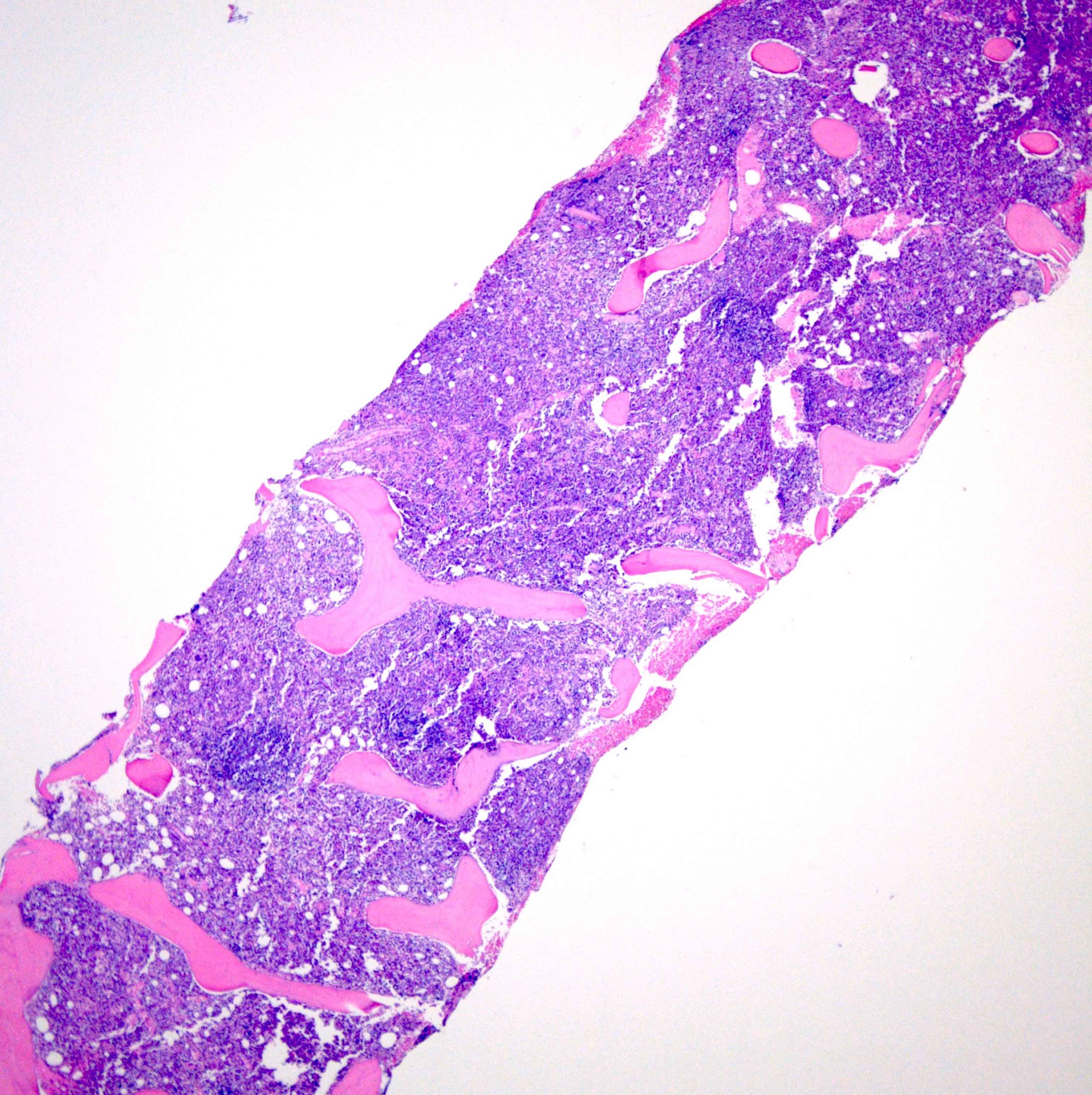
Markedly hypercellular bone marrow
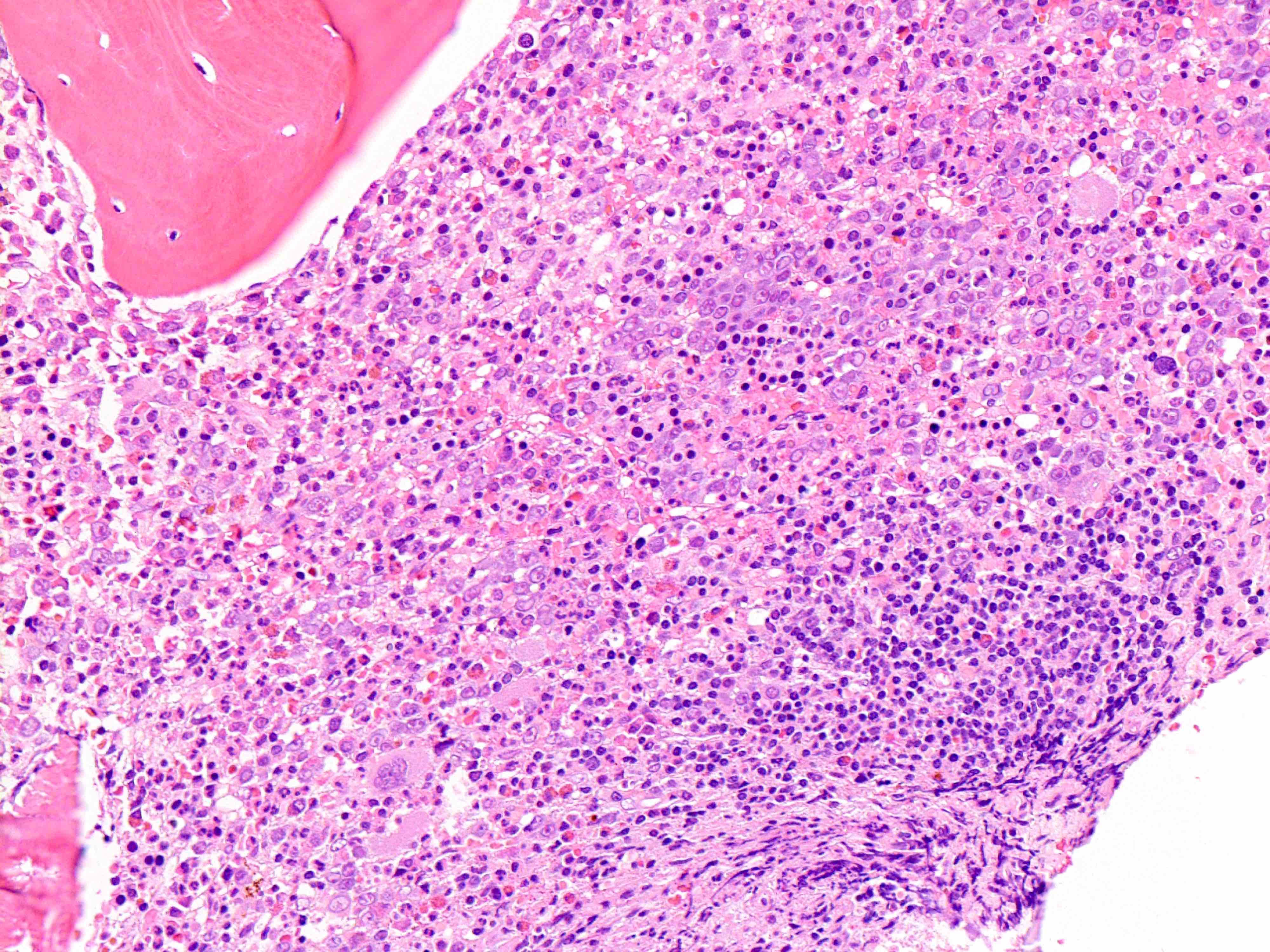
Erythroid hyperplasia
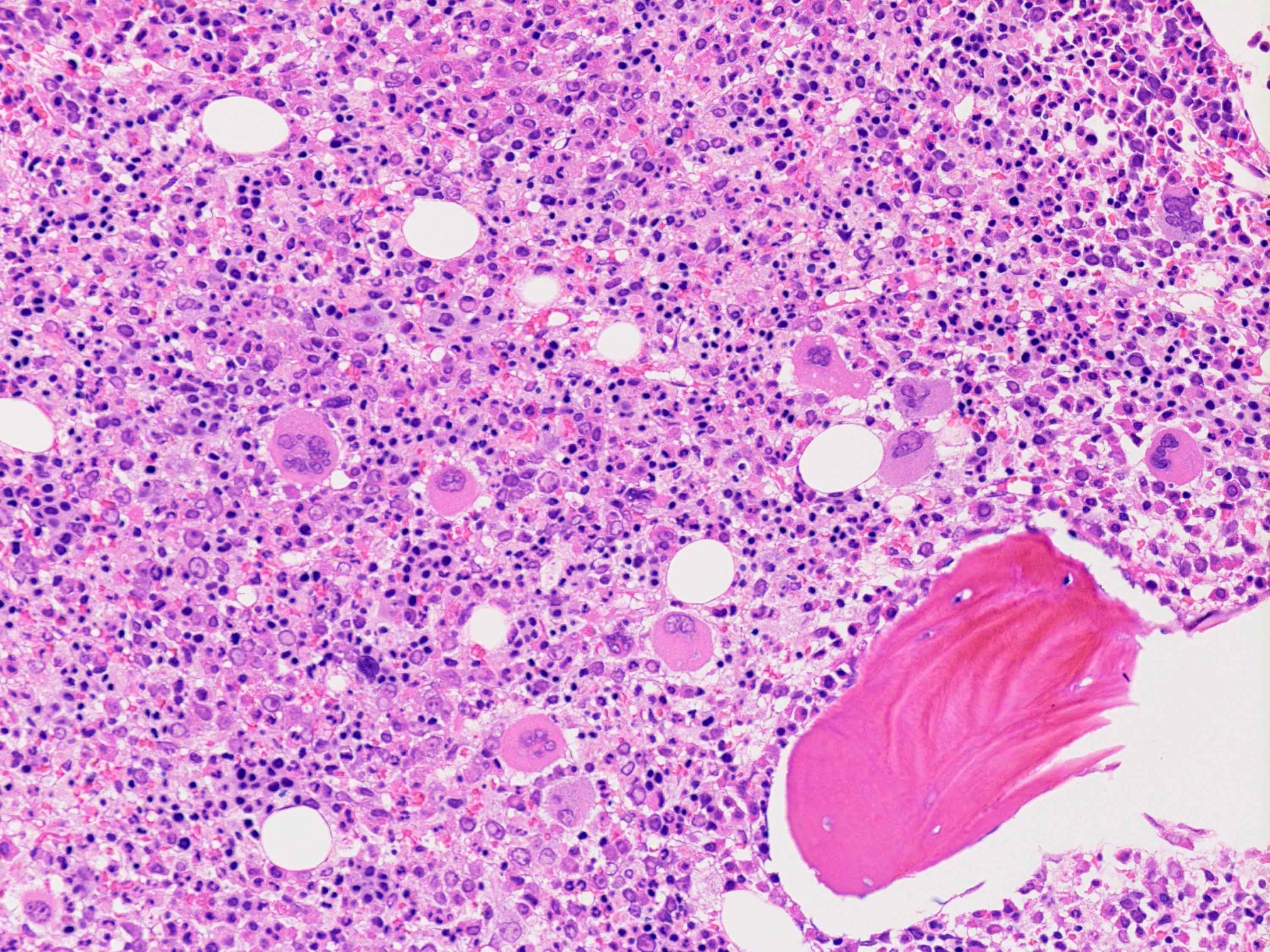
Megakaryocytic hyperplasia without atypia
Sinusoidal dilation
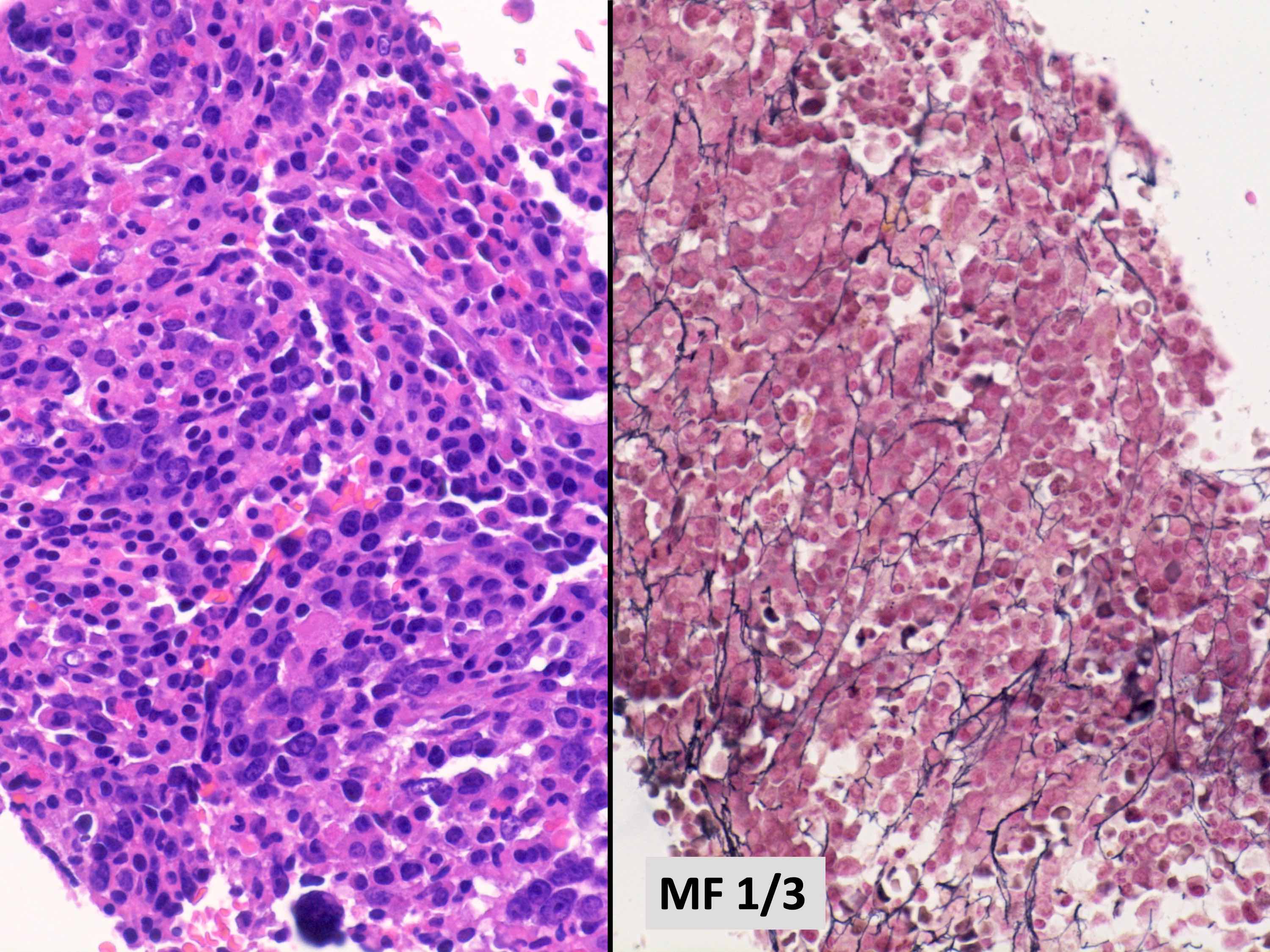
MF grade 1
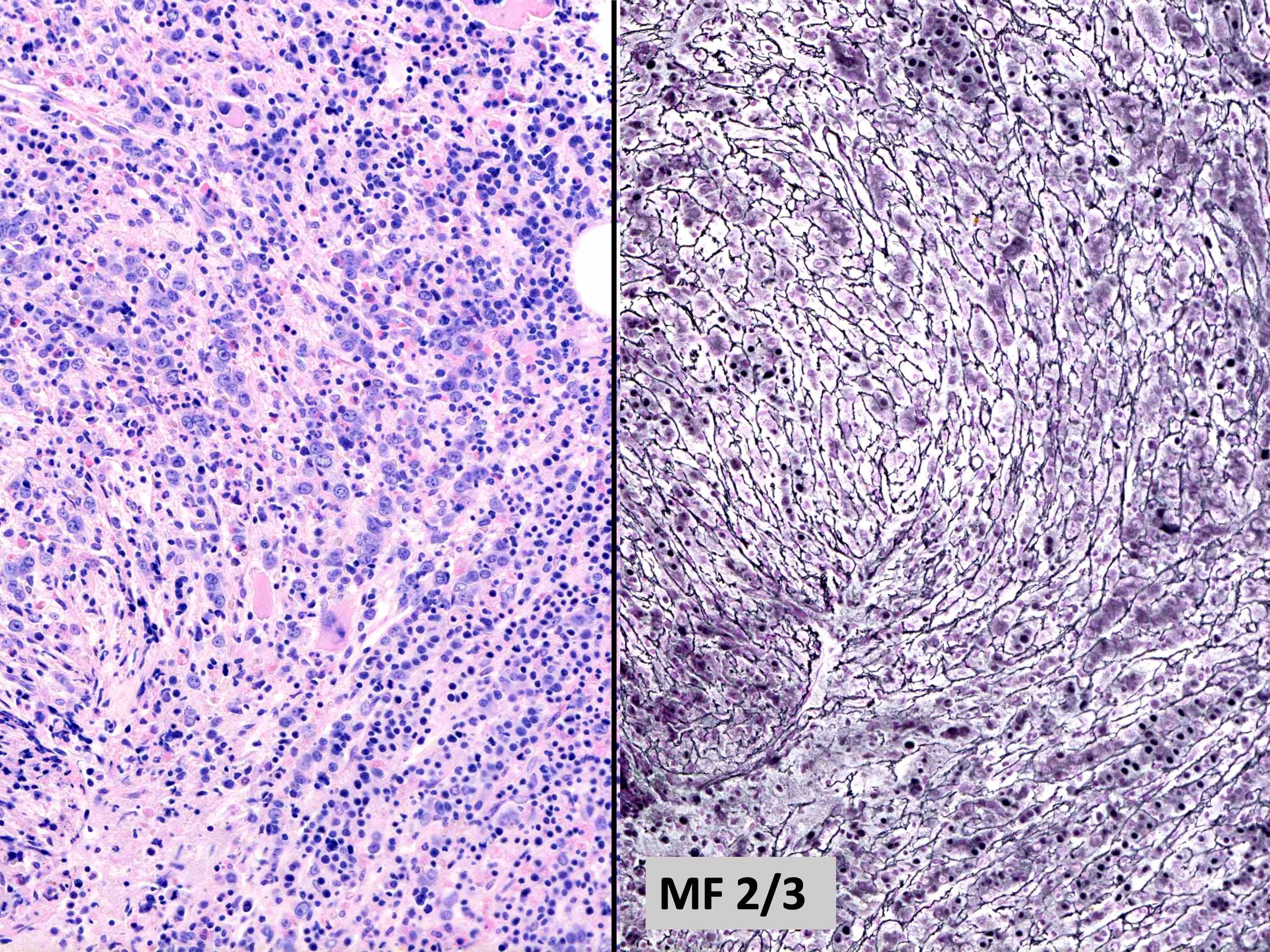
MF grade 2
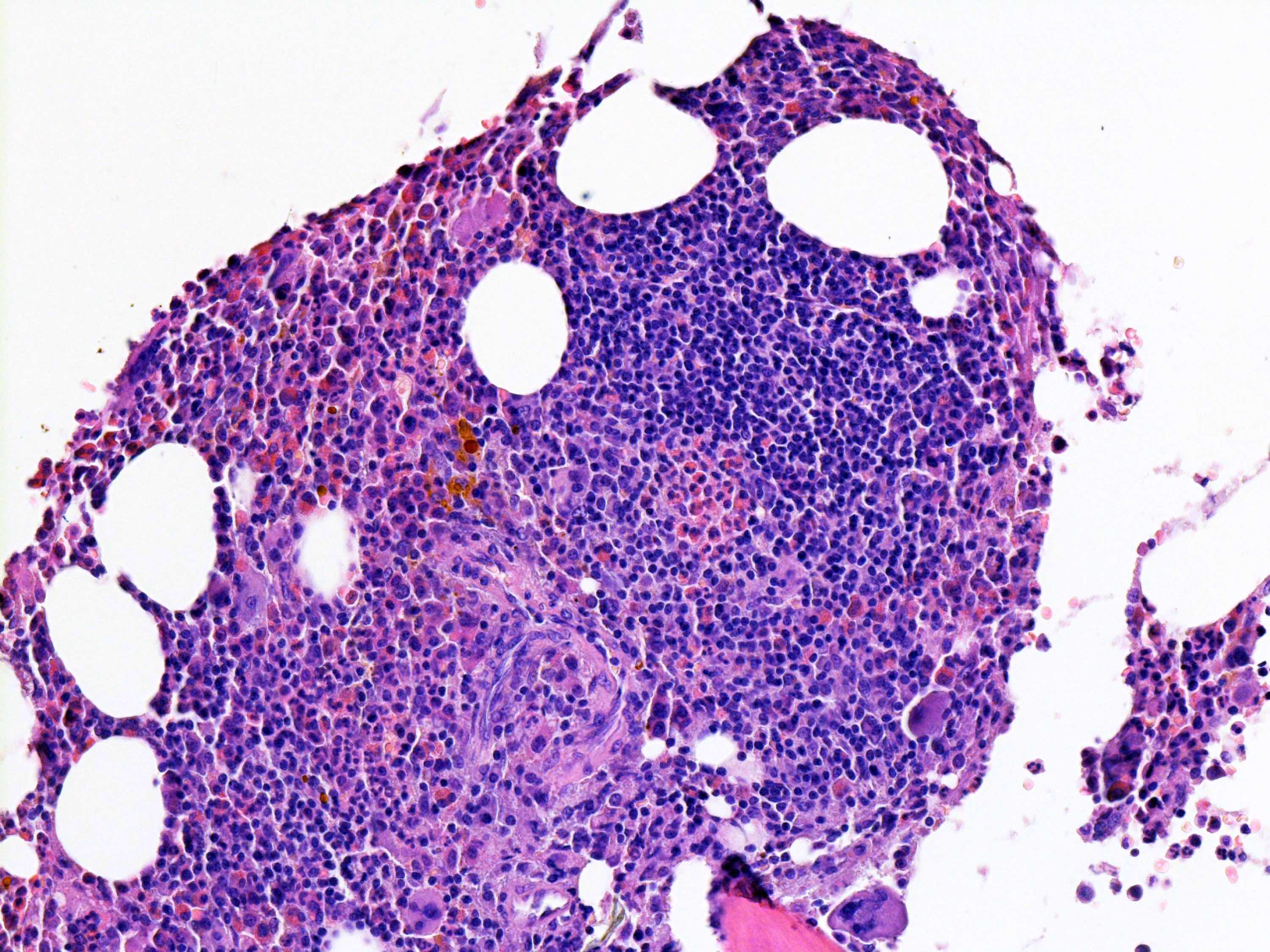
Benign lymphoid aggregate
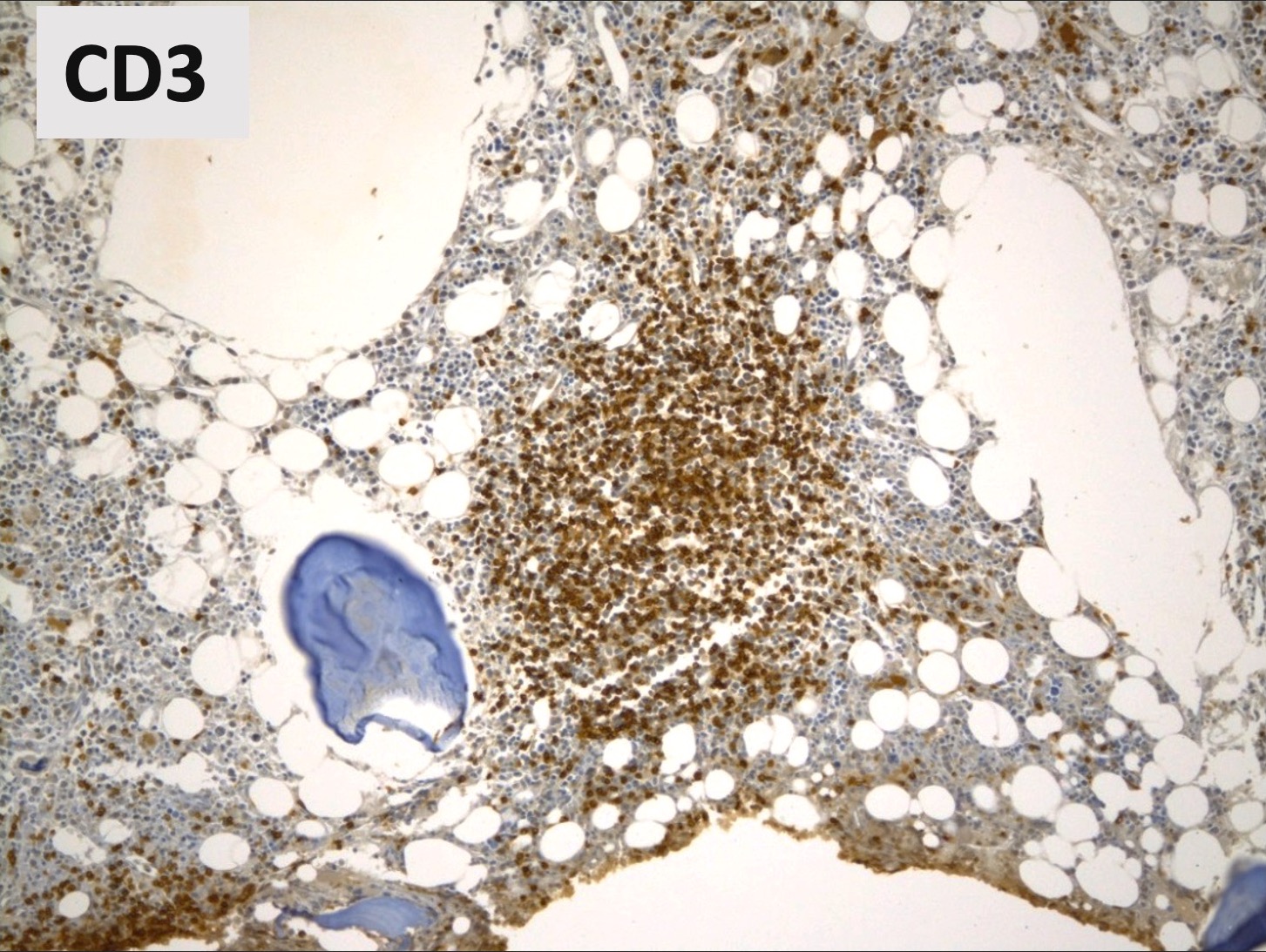
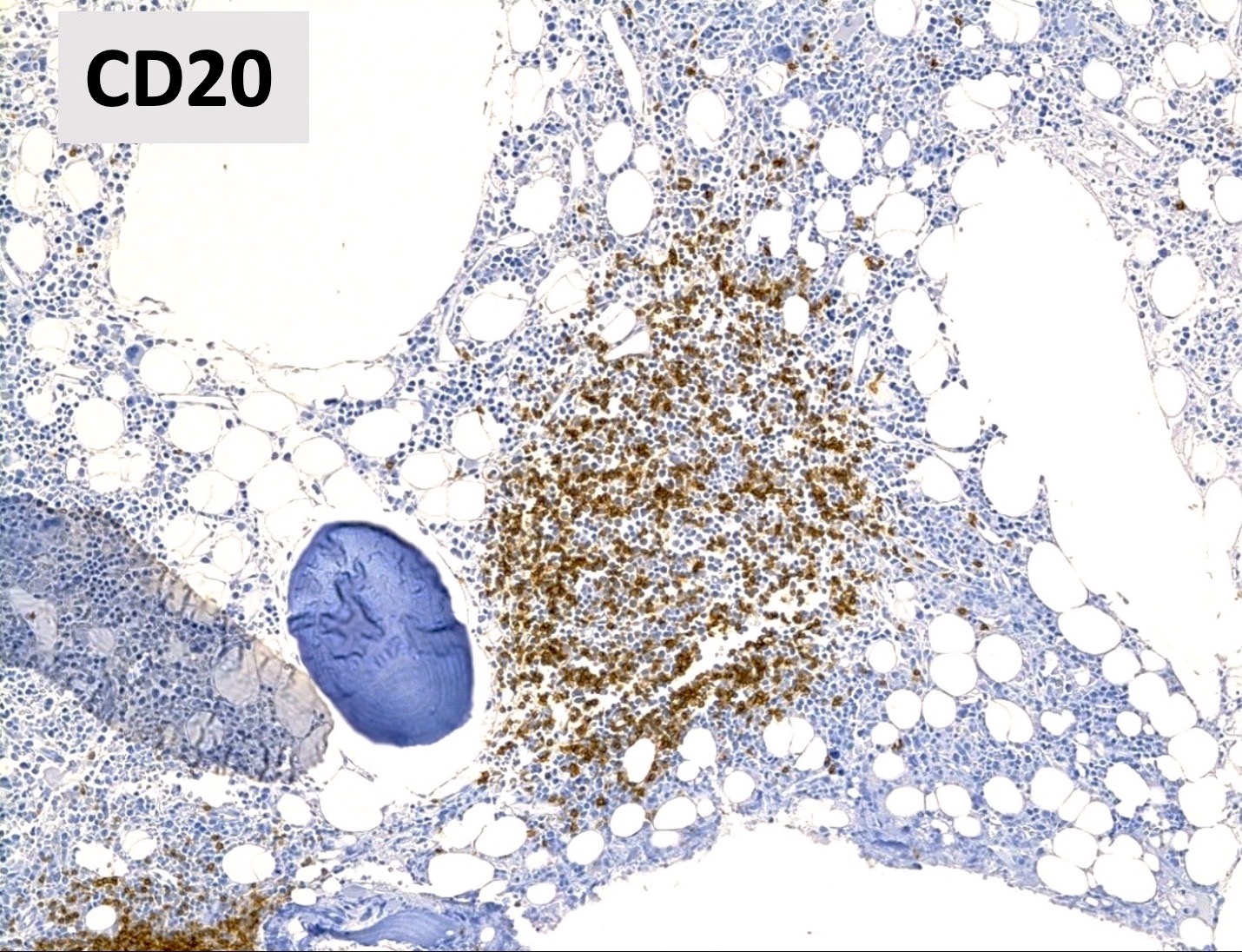
Benign lymphoid aggregate immunostains
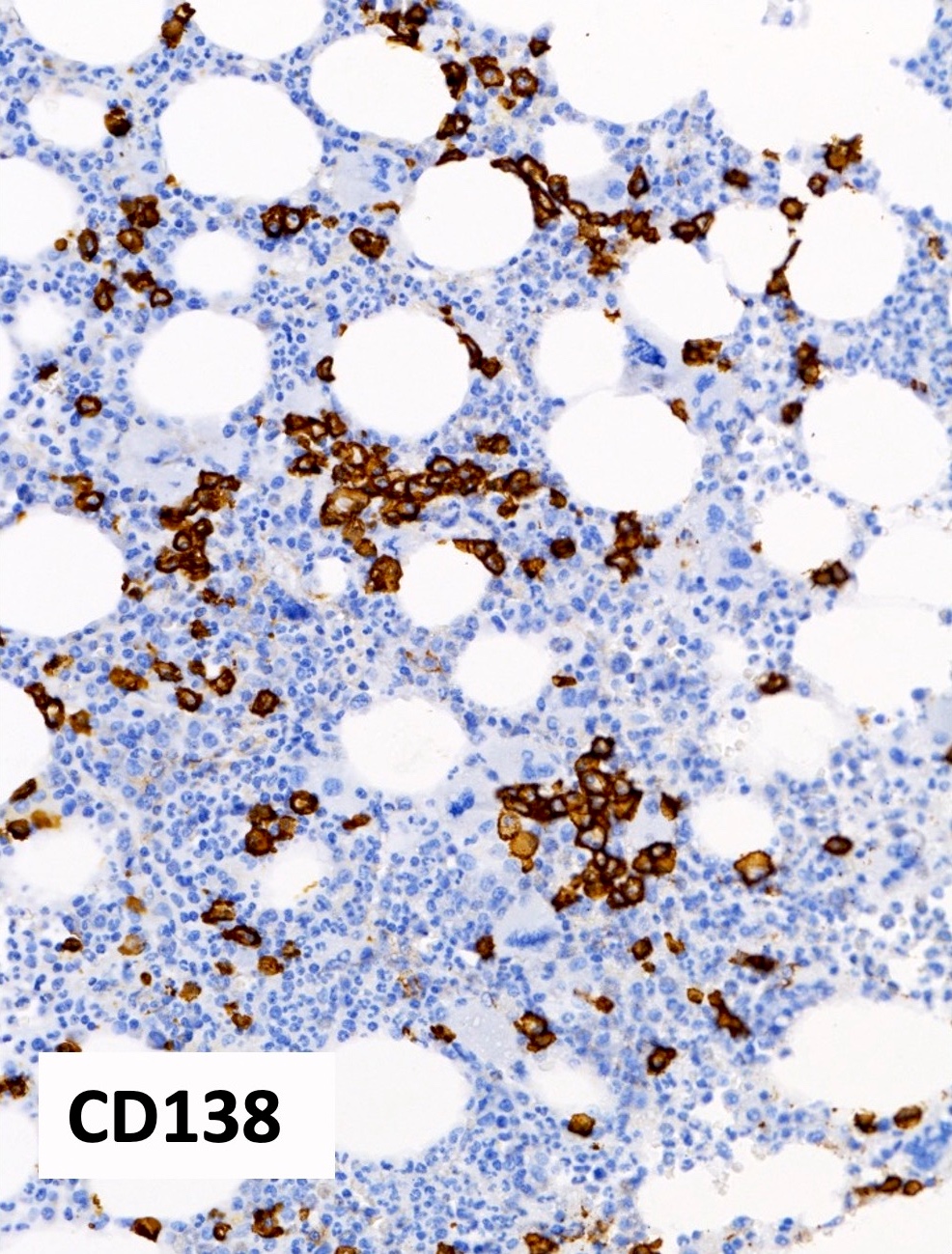

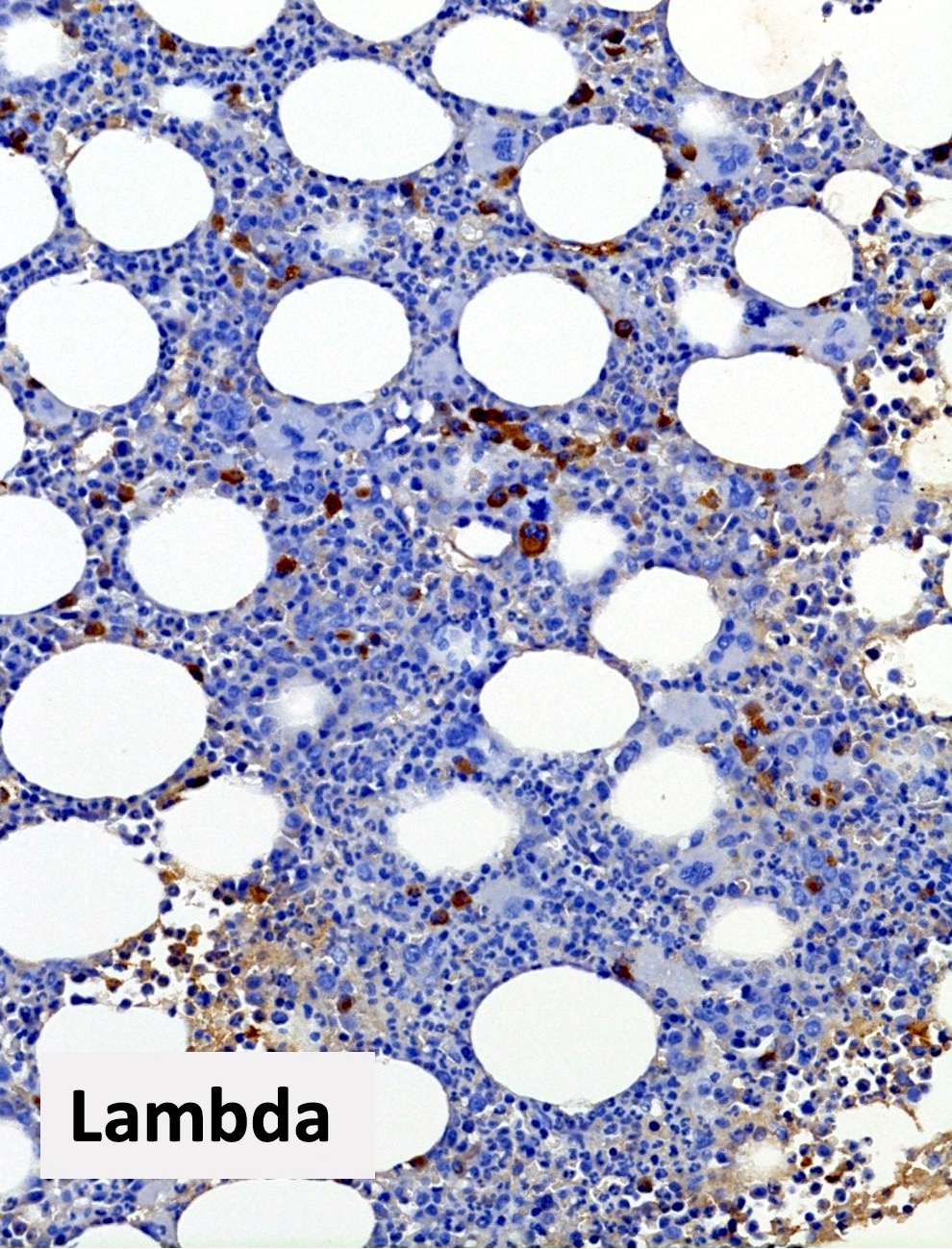
Polytypic plasmacytosis
Peripheral smear description
- Anemia, thrombocytopenia and / or leukopenia
- No or rare dacrocytes (teardrop cells)
- No or rare nucleated red blood cells and blasts
- Usually no eosinophilia or basophilia
- No dysplasia (i.e., no pseudo Pelger-Huët changes, neutrophilic hypogranularity)
- No features worrisome for a myeloid neoplasm (e.g., no significant increase in basophils, eosinophils, neutrophilic left shift, circulating megakaryocytes, etc.)
- Reference: Hum Pathol 2014;45:2183
Peripheral smear images
Contributed by Ria Vergara-Lluri, M.D.
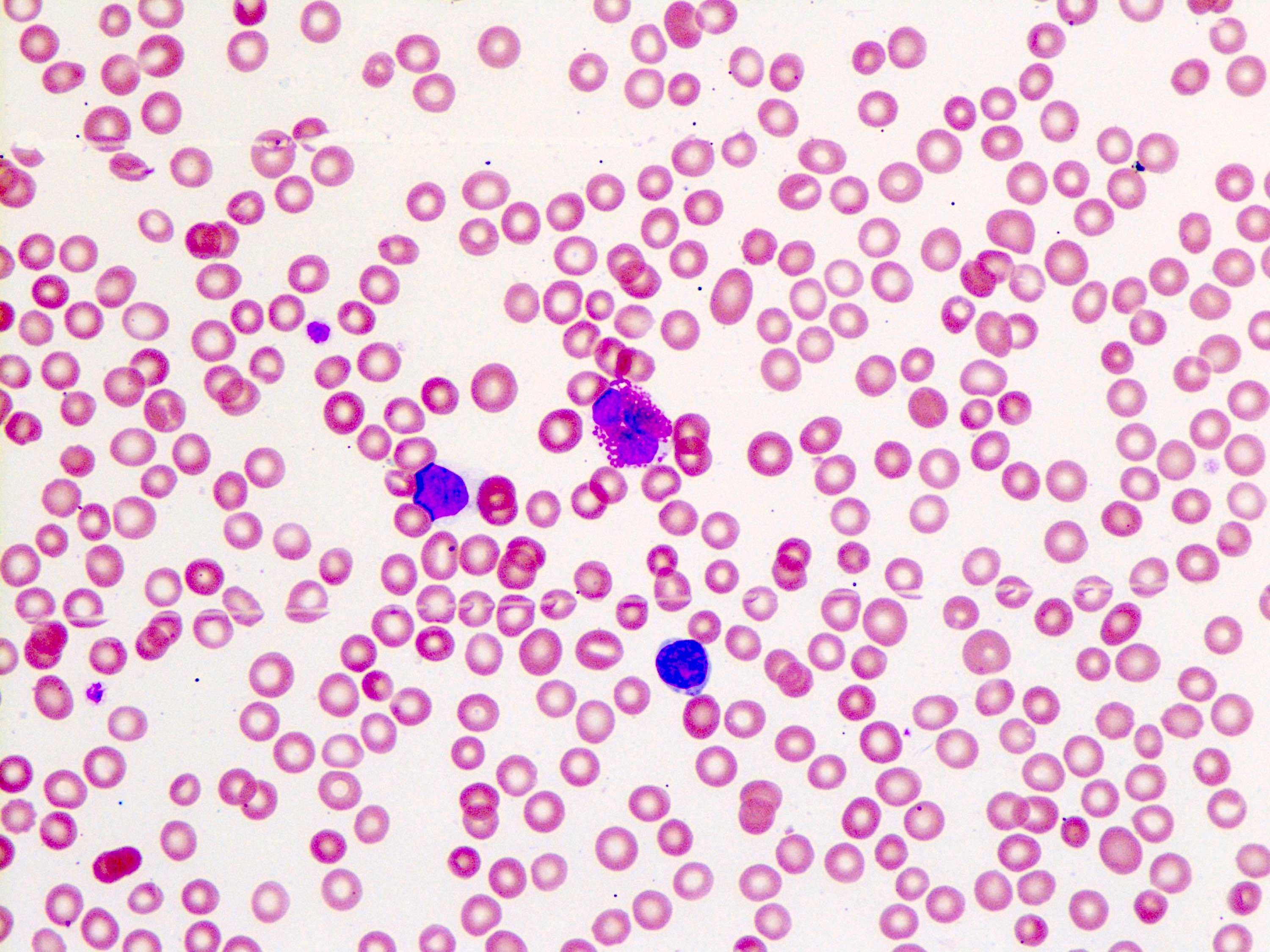
Peripheral blood smear
Positive stains
- Reticulin: highlights bone marrow stromal fibrosis
- CD3 / CD20
- Benign lymphoid aggregates display reactive patterns
- Predominance of T cells over B cells; core of many T cells with peripheral rim of B cells or mixture of T and B cells (Hum Pathol 2013;44:512)
- Interstitial infiltrates show predominance of T over B cells (Pediatr Blood Cancer 2023;70:e30144)
- Benign lymphoid aggregates display reactive patterns
- CD138 / kappa / lambda: show mild polytypic plasmacytosis, if present
- Reference: Hum Pathol 2014;45:2183
Flow cytometry description
- No overt myeloid dysmaturation
- No excess blasts (typically < 3%)
- Polytypic B cells with normal kappa to lambda ratios (Hum Pathol 2014;45:2183)
- No overt pan T cell aberrancies
Molecular / cytogenetics description
- Role of potential undiscovered driver mutations is unknown (Haematologica 2021;106:871)
- 1 pediatric AIMF case series found inborn errors of immunity in 50% of cases (n=8) (Pediatr Blood Cancer 2023;70:e30144)
- No JAK2, CALR, MPL mutations
- No molecular or cytogenetic markers of other clonal disease (Eur J Haematol 2023;111:706)
- Negative molecular / cytogenetic testing has been suggested as diagnostic criterion (Haematologica 2021;106:871)
Sample pathology report
- Peripheral blood:
- Mild normochromic, normocytic anemia
- No circulating blasts
- Mild neutropenia with no overt dysplastic features
- No lymphocytosis nor significant increase in large granular lymphocytes
- Mild thrombocytopenia
- Bone marrow aspirates, imprints, trephine and clot biopsies:
- Moderately hypercellular bone marrow with moderate erythroid and megakaryocytic hyperplasias and adequate numbers of granulocytic precursors
- Mild reticulin fibrosis (MF grade 1; scale 0 - 3)
- Mild lymphocytosis with benign lymphoid aggregates
- Mild polytypic plasmacytosis
- No excess blasts (blasts < 5%)
- No overt dysplastic features
- No diagnostic evidence of lymphoma or plasma cell dyscrasia
- Comment: The constellation of morphologic features is not specific for any one etiology and can also be found in other nonneoplastic settings (such as in other inflammatory disorders, infections, drug exposures, etc.). Thus, these features must be interpreted in the context of pertinent clinical information (e.g., presence of an established rheumatologic disease, etc.), laboratory studies (e.g., rheumatoid factor, antineutrophil antibodies, direct antiglobulin tests, etc.) and imaging findings (e.g., evaluation for splenomegaly, lymphadenopathy, etc.) and may best be undertaken with consultation / communication with hematology and rheumatology clinical services. If other etiologies have been adequately excluded and in the appropriate clinicopathologic context, these morphologic findings could be compatible with autoimmune myelofibrosis (AIMF).
Differential diagnosis
- Nonneoplastic myelofibrosis due to other causes (e.g., idiopathic multicentric Castleman disease, drug induced, infection) (Hematol Oncol 2022;40:191, Bain: Bone Marrow Pathology, 5th Edition, 2019, Br J Haematol 2007;139:351):
- Clinical history or laboratory studies consistent with alternate cause
- For example, idiopathic multicentric Castleman disease (iMCD) can show similar morphologic findings (such as hypercellularity, marrow reticulin fibrosis and megakaryocytic hyperplasia)
- For example, recent romiplostim or eltrombopag administration; evidence of osteomyelitis, tuberculosis or leishmaniasis
- Essential to thoroughly integrate data from clinical history, physical examination, other pathology (e.g., lymph node biopsy results) and imaging findings
- Clinical history or laboratory studies consistent with alternate cause
- Primary myelofibrosis (PMF), prefibrotic and overt fibrotic stages (Acta Haematol 2017;138:129):
- Myeloproliferative neoplasm with poor prognosis
- Palpable splenomegaly (~89 - 100%) and hepatomegaly (~54 - 89%) (Medicine (Baltimore) 1983;62:353)
- May have positive autoimmune antibody serologies (Clin Adv Hematol Oncol 2018;16:619)
- Bone marrow core biopsy
- Prefibrotic stage
- Increased myeloid:erythroid ratio and / or trilineage expansions
- No to minimal reticulin fibrosis (MF0 - MF1)
- Atypical megakaryocyte proliferation
- Often bizarre, hyperchromatic nuclei distributed in loose or dense clusters
- Overt fibrotic stage
- Similar to prefibrotic stage, although cases with more advanced fibrosis may show hypocellularity and / or cellular dropout
- Moderate to severe reticulin fibrosis (MF2 to MF3)
- Varying degrees of osteosclerosis
- Prefibrotic stage
- Driver mutations (JAK2, CALR or MPL) or other clonal markers / cytogenetic abnormalities usually present
- Post myeloproliferative neoplasm myelofibrosis (e.g., post polycythemia vera or post essential thrombocythemia myelofibrosis):
- Prior documented diagnosis of polycythemia vera or essential thrombocythemia
- May present with increasing splenomegaly
- May have positive autoimmune antibody serologies (Acta Haematol 2017;138:129)
- Bone marrow core biopsy
- Bone marrow reticulin fibrosis MF2 or MF3
- Clusters of atypical megakaryocytes
- May show osteosclerosis
- Often has JAK2, CALR or MPL mutation
- Non-Hodgkin lymphomas:
- Chronic myelomonocytic leukemia (CMML) with fibrosis (Oncotarget 2017;8:103274):
- May have splenomegaly (58%, n=45)
- Persistent peripheral blood monocytosis (≥ 1 x 109/L)
- Monocytes > 10% of peripheral blood leukocytes
- Will have morphologic dysplasia in ≥ 1 hematopoietic lineage
- Will have molecular / cytogenetic abnormalities
- Desmoplasia associated with metastatic tumors (Bain: Bone Marrow Pathology, 5th Edition, 2019):
- Normochromic, normocytic anemia is common
- Other cytopenias are less common
- May see leukoerythroblastic anemia (< 50%)
- Aspirate may show tumor cells, necrosis, osteoblasts / osteoclasts
- Trephine may also show osteolysis / osteosclerosis, neoangiogenesis
- Normochromic, normocytic anemia is common
Additional references
Board review style question #1
A 47 year old woman presents with fatigue, joint pains and pallor. Laboratory workup is notable for severe anemia, thrombocytopenia and a serum ANA titer of 1:40. A bone marrow biopsy is obtained and shows marked hypercellularity with scattered nonparatrabecular lymphoid aggregates (images above) as well as myelofibrosis highlighted by reticulin staining (not shown). Which of the following findings would be typical for a diagnosis of autoimmune myelofibrosis (AIMF)?
- Increased numbers of megakaryocytes in dense clusters
- MPL mutation on next generation sequencing (NGS) myeloid panel
- Palpable splenomegaly
- Response to glucocorticoids
- Severe reticulin fibrosis (MF grade 3)
Board review style answer #1
D. Response to glucocorticoids. Autoimmune myelofibrosis (AIMF) is a nonneoplastic condition that may be responsive to a course of glucocorticoids. Overall, the diagnosis of AIMF requires careful correlation of the patient's clinical information with the histopathologic findings on bone marrow biopsy. By morphology, AIMF is associated with a hypercellular marrow (typically with erythroid and megakaryocytic hyperplasias), benign lymphoid aggregates (e.g., nonparatrabecular, polyclonal lymphocytes), polytypic plasmacytosis and without overt features of dysplasia.
Answer E is incorrect because myelofibrosis in AIMF is usually mild to moderate, although rare cases of AIMF have been associated with severe reticulin fibrosis and this finding alone would not be inconsistent with the diagnosis. Answer C is incorrect because palpable splenomegaly is quite uncommon in AIMF, though not exclusionary. Answer B is incorrect because the presence of a pathogenic mutation in MPL ought to prompt assessment favoring neoplasia. The absence of somatic mutations on next generation sequencing panels for myeloid malignancies (e.g., JAK2, CALR and MPL) can be an essential part of the diagnostic work up of AIMF. Answer A is incorrect because while it is typical to see increased numbers of megakaryocytes in AIMF, dense clusters of megakaryocytes (which are very likely to also display cytologic atypia) would be highly unusual for (and morphologically inconsistent with) AIMF. This observation strongly favors a myeloid neoplasm (such as primary myelofibrosis) and should trigger a thorough work up to demonstrate clonality (e.g., karyotype, NGS testing).
Comment Here
Reference: Autoimmune myelofibrosis (AIMF)
Answer E is incorrect because myelofibrosis in AIMF is usually mild to moderate, although rare cases of AIMF have been associated with severe reticulin fibrosis and this finding alone would not be inconsistent with the diagnosis. Answer C is incorrect because palpable splenomegaly is quite uncommon in AIMF, though not exclusionary. Answer B is incorrect because the presence of a pathogenic mutation in MPL ought to prompt assessment favoring neoplasia. The absence of somatic mutations on next generation sequencing panels for myeloid malignancies (e.g., JAK2, CALR and MPL) can be an essential part of the diagnostic work up of AIMF. Answer A is incorrect because while it is typical to see increased numbers of megakaryocytes in AIMF, dense clusters of megakaryocytes (which are very likely to also display cytologic atypia) would be highly unusual for (and morphologically inconsistent with) AIMF. This observation strongly favors a myeloid neoplasm (such as primary myelofibrosis) and should trigger a thorough work up to demonstrate clonality (e.g., karyotype, NGS testing).
Comment Here
Reference: Autoimmune myelofibrosis (AIMF)
Board review style question #2
A 37 year old woman with no significant medical history was admitted to the hospital for acute onset of fevers and severe fatigue. Her vital signs and physical examination were significant only for fever and tachycardia; there is no palpable hepatosplenomegaly.
- Imaging studies showed no organomegaly or lymphadenopathy
- Laboratory studies were remarkable for WBC 3.1 x 109/L, Hb 8.0 g/dL, Plt 39 x 103/uL, ANA titer of 1:80 and a negative DAT
- Peripheral blood smear showed cytopenias without leukoerythroblastosis
- Remainder of the infectious, autoimmune and neoplastic workup was negative
- Bone marrow biopsy displayed a markedly hypercellular marrow with erythroid and megakaryocytic hyperplasias, scattered nonparatrabecular lymphoid aggregates, no overt dysplasia or excess blasts and mild polytypic plasmacytosis (< 10%)
- Reticulin staining revealed moderate reticulin fibrosis (MF2; image shown above)
- Normal karyotype was reported while next generation sequencing (NGS) myeloid panels for somatic mutations were negative
She received a course of glucocorticoids and achieved complete hematologic response and was discharged from the hospital. Rheumatology consultation revealed no established autoimmune disorder. She is being closely monitored by the hematology clinical service. A repeat biopsy performed 1 year after her hospital admission showed similar findings, though decreased reticulin fibrosis is observed (MF1). Which of the following is the most appropriate diagnosis at this time?
- Myelofibrosis, secondary to polycythemia vera
- Primary autoimmune myelofibrosis
- Primary myelofibrosis, overt stage
- Primary myelofibrosis, prefibrotic stage
Board review style answer #2
B. Primary autoimmune myelofibrosis. Autoimmune myelofibrosis (AIMF) is a nonneoplastic condition that may occur in the presence of another predefined autoimmune disorder (secondary AIMF) or in the presence of nonspecific autoantibodies / symptoms not meeting criteria for another established autoimmune disorder (primary AIMF).
Secondary AIMF has been described in association with many autoimmune disorders, including systemic lupus erythematosus, rheumatoid arthritis, Sjögren syndrome, autoimmune hemolytic anemia and others. In this vignette, over the course of close clinical follow up, no specific autoimmune, infectious / inflammatory or other hematologic disorder could be established. Furthermore, her favorable response to glucocorticoids and mild decrease in reticulin fibrosis on repeat biopsy is typical of AIMF. Thus, a diagnosis of primary AIMF may be suggested at this time and is the most appropriate choice of those listed here, after careful consideration of the bone marrow findings in the context of clinical information and disease course. Alternate causes of myelofibrosis (including, for example, exposure to drugs, infectious causes, myeloproliferative neoplasm (MPN) or post-MPN myelofibrosis) must be ruled out. Answer A is incorrect because in this case, the patient described has no significant medical history nor evidence of neoplastic progression, making post-MPN myelofibrosis unlikely. Answers C and D are incorrect because the vignette describes a characteristic combination of morphologic and genetic findings that would be seen with AIMF, including the absence of somatic mutations on next generation sequencing panels for myeloid malignancies (e.g., JAK2, CALR and MPL), which can be an essential part of the diagnostic work up.
Comment Here
Reference: Autoimmune myelofibrosis (AIMF)
Secondary AIMF has been described in association with many autoimmune disorders, including systemic lupus erythematosus, rheumatoid arthritis, Sjögren syndrome, autoimmune hemolytic anemia and others. In this vignette, over the course of close clinical follow up, no specific autoimmune, infectious / inflammatory or other hematologic disorder could be established. Furthermore, her favorable response to glucocorticoids and mild decrease in reticulin fibrosis on repeat biopsy is typical of AIMF. Thus, a diagnosis of primary AIMF may be suggested at this time and is the most appropriate choice of those listed here, after careful consideration of the bone marrow findings in the context of clinical information and disease course. Alternate causes of myelofibrosis (including, for example, exposure to drugs, infectious causes, myeloproliferative neoplasm (MPN) or post-MPN myelofibrosis) must be ruled out. Answer A is incorrect because in this case, the patient described has no significant medical history nor evidence of neoplastic progression, making post-MPN myelofibrosis unlikely. Answers C and D are incorrect because the vignette describes a characteristic combination of morphologic and genetic findings that would be seen with AIMF, including the absence of somatic mutations on next generation sequencing panels for myeloid malignancies (e.g., JAK2, CALR and MPL), which can be an essential part of the diagnostic work up.
Comment Here
Reference: Autoimmune myelofibrosis (AIMF)
