
Liver & intrahepatic bile ducts
Developmental anomalies / cysts
Progressive familial intrahepatic cholestasis
Authors: Puja Sakhuja, M.D., Surbhi Goyal, M.D.
Editorial Board Member: Aaron R. Huber, D.O.
Deputy Editor-in-Chief: Catherine E. Hagen, M.D.


Last author update: 15 June 2021
Last staff update: 15 June 2021
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: Progressive familial intrahepatic cholestasis[TI] pathology
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Immunohistochemistry & special stains | Electron microscopy description | Electron microscopy images | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Sakhuja P, Goyal S. Progressive familial intrahepatic cholestasis. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/liverpfic.html. Accessed April 26th, 2024.
Definition / general
- Progressive familial intrahepatic cholestasis (PFIC) is a type of neonatal cholestatic liver disease resulting from an inherited molecular defect
Essential features
- Autosomal recessive hereditary liver disorders
- Presents in infancy or childhood with intrahepatic cholestasis
- Progresses to liver failure in the first decade of life
- Defective bile secretion and metabolism
Terminology
- Byler disease / PFIC1 / FIC1 deficiency
- Byler syndrome / PFIC2 / BSEP deficiency
- PFIC3 / MDR3 deficiency
- Types:
- PFIC1
- PFIC2
- PFIC3
- PFIC4
- PFIC5
- PFIC6
ICD coding
- ICD-10: P78.89 - other specified perinatal digestive system disorders
Epidemiology
- Comprises 10 - 15% of cases of pediatric cholestatic liver disease
- Incidence is between 1:50,000 - 1:100,000 births (Clin Res Hepatol Gastroenterol 2012;36:S26)
- No sex or geographical predilection
Sites
- Liver
Pathophysiology
- Defective bile acid homeostasis, transport or excretion
- Injury to bile epithelium or canalicular membrane due to high bile salts
- PFIC1:
- FIC1 protects the canalicular membrane from the detergent action of high bile salt concentrations by flipping the cell membrane phospholipids
- Lower expression of FIC1 gene causes downregulation of the nuclear receptor farnesoid X, leading to upregulation of bile acid synthesis, increased bile acid reabsorption in intestine and secondary reduction in the expression of bile salt export pump (BSEP)
- Accumulation of intracellular bile salts coupled with the absence of the protective lipid flipping damages the hepatocytes (Dig Liver Dis 2019;51:922)
- FIC1 is expressed in intestines, pancreas and kidney
- PFIC2:
- Mutations in ABCB11 cause downregulation of the ATP dependent BSEP located at the hepatocyte canalicular membrane
- Reduced pumping of bile salts out of hepatocytes (Semin Liver Dis 2011;31:3)
- PFIC3:
- Mutations in class III multidrug resistance p-glycoprotein (MDR3) result in defective transport of phosphatidylcholine across the canaliculus
- High bile salt to phospholipid ratio with hydrophobic bile acids injure the cholangiocytes and result in cholangitis
- Cholesterol can crystallize into stones, leading to cholelithiasis
- PFIC4:
- Tight junction protein 2 (TJP2) protein is one of the intracellular anchors for tight junctions that seal canaliculi and prevent damage from cytotoxic detergent bile salts
- Loss of function of TJP2 causes bile leakage into the hepatocytes and elicits inflammatory response and fibrosis
- PFIC5:
- Farnesoid X receptor (FXR) is a bile acid activated nuclear transcription factor that regulates bile acid metabolism by inducing the expression of BSEP and MDR3
- Loss of FXR leads to increased bile acid synthesis and severe neonatal cholestatic disease
- PFIC6:
- Associated with mutation in the MYO5B gene
- MYO5B interacts with RAB11A to facilitate normal trafficking of ABC transporter proteins, including BSEP, to the canalicular membrane
- MYO5B plays a role in plasma membrane recycling and epithelial cell polarization in enterocytes and respiratory epithelial cells
Etiology
- Inherited genetic defects as described in pathophysiology
Diagrams / tables
Images hosted on other servers:

Types of PFIC
Clinical features
- Unremitting or persistent cholestasis with jaundice and intractable pruritus
- PFIC1:
- Onset in first 3 - 6 months of life
- Diarrhea with malabsorption of fats and fat soluble vitamins leads to coagulopathy and rickets
- Extrahepatic manifestations include sensorineural hearing loss, persistent diarrhea, cholecystitis, pancreatitis, failure to thrive, short stature and respiratory symptoms (World J Hepatol 2019;11:450)
- Benign recurrent intrahepatic cholestasis (BRIC) patients have milder phenotype with self remitting episodic cholestasis
- PFIC2:
- Onset from birth to 6 months of life
- Liver specific disease
- No extrahepatic manifestations
- PFIC3:
- Presents in late infancy to young adulthood
- Variable jaundice, hepatomegaly and acholic stools
- 33% of cases present within the first year of life
- May manifest as adult biliary cirrhosis, low phospholipids cholelithiasis, intrahepatic cholestasis of pregnancy, transient neonatal cholestasis and drug induced cholestasis
- Less severe, with slow evolution to cirrhosis
- PFIC4:
- Complex genotype - phenotype presentations, which are age dependent and variable penetrance
- Affects children and young adults
- Presents as intrahepatic cholestasis of pregnancy, pruritis, portal hypertension and progression to biliary cirrhosis
- Extrahepatic features, such as hearing loss and respiratory symptoms, seen
- PFIC5:
- Very rare, about 8 cases reported in literature (World J Gastroenterol 2020;26:7470)
- Neonatal onset of normal gamma glutamyl transferase (GGT) associated cholestasis
- Early onset vitamin K independent coagulopathy and markedly elevated AFP levels are relatively specific (Nat Commun 2016;7:10713)
- Rapid progression into micronodular cirrhosis
- PFIC6:
- Low to normal GGT levels, jaundice, pruritus in pediatric patients
Diagnosis
- Correlation of clinical presentation with imaging, biochemical, histopathological, ultrastructural and genetic findings
Laboratory
- Conjugated hyperbilirubinemia
- Raised serum alkaline phosphatase
- Gamma glutamyl transferase: normal or low in all types of PFIC except type 3 (raised to over 10 times the normal levels)
- Elevated alanine or aspartate aminotransferase levels (ALT or AST) > 2 times normal limits suggests PFIC2 (J Hepatol 2010;53:170)
- Coagulation profile: deranged in PFIC1 and 5
- Serum alphaprotein levels: elevated in PFIC2 and 5
- Serum bile acids: elevated
- Bile analysis: technically demanding but helpful in exact subtyping
- Low bile acids with normal phospholipids seen in PFIC1 and 2
- Phospholipids are markedly reduced in PFIC3 (< 15% of total biliary lipids; normal: 19 - 24%) (J Clin Exp Hepatol 2014;4:25)
Radiology description
- Ultrasound:
- Normal or marked enlargement in gallbladder size with presence of stones can be seen in PFIC2 and 3
- Rules out extrahepatic causes of obstructive jaundice, like extrahepatic biliary atresia (EHBA), choledochal cysts, tumor and lymphadenopathy (J Pediatr Surg 1999;34:1706)
- Cholangiography:
- Can confirm EHBA, rule out sclerosing cholangitis and can be used for sampling bile
Prognostic factors
- PFIC1:
- Variable presence of cirrhosis is poor prognostic factor requiring liver transplant
- Extrahepatic symptoms, like diarrhea and fat malabsorption, persist or worsen after liver transplantation
- Steatohepatitis has been reported in liver allografts (Hepatol Commun 2018;2:515)
- PFIC2:
- More severe, with rapid progression to end stage liver disease in initial few years
- Association with hepatocellular carcinoma (15%), cholangiocarcinoma and pancreatic tumors (Hepatology 2006;44:478, World J Hepatol 2019;11:450)
- Development of BSEP specific alloreactive antibodies, leading to recurrence in liver transplantation patients
- E297G / D482G mutations in ABCB11 gene associated with milder and slower disease progression and good response to biliary diversion procedures (Hepatol Commun 2018;2:515)
- PFIC3:
- Hepatocellular carcinoma and cholangiocarcinoma have been associated with PFIC3 (J Clin Exp Hepatol 2014;4:25)
- Biliary diversion procedures are not as effective but liver transplantation is curative
- PFIC4:
- Can present with rapidly progressive cirrhosis and hepatocellular carcinoma during adolescence
- Liver transplantation is successful
- PFIC5:
- Rapid progression to cirrhosis and liver failure in the first 2 years of life with worsening coagulopathy (Nat Commun 2016;7:10713)
- Liver transplantation is successful in few cases
- PFIC6:
- MYO5B defects associated with microvillous inclusion disease (MVID) and diarrhea (Hepatology 2014;60:301)
- Lifelong total parenteral nutrition (TPN) is required but associated with increased risk of sepsis and small bowel transplant
Case reports
- 2 month old girl presented with jaundice since 1 month (World J Gastroenterol 2009;15:4339)
- 4 month old girl with severe jaundice, pruritus and clay colored stools (Medicine (Baltimore) 2019;98:e15593)
- 7 month old girl with cholestasis since last 6 months (Pediatr Gastroenterol Hepatol Nutr 2019;22:479)
- 8 month old girl presented with acute liver cell failure (Indian J Pathol Microbiol 2010;53:334)
- 7 year boy with persistent jaundice (Saudi J Gastroenterol 2011;17:212)
- 19 year man with cirrhosis and variceal bleeding (World J Gastroenterol 2020;26:550)
Treatment
- Choleretics, such as ursodeoxycholic acid (UDCA), are the first line medical treatment in all types of PFIC, hydrophilic bile acid that induces the expression of BSEP and MDR3 (World J Hepatol 2019;11:450)
- 4 phenylbutyrate increases cell surface expression of the mutant BSEPs and improves liver function and pruritus in PFIC2 (J Clin Exp Hepatol 2014;4:25)
- Chemical chaperones can improve MDR3 trafficking in PFIC3
- Biliary diversion effective in 80% of patients (J Clin Exp Hepatol 2014;4:25)
- Liver transplantation is the last resort in patients who develop cirrhosis and liver failure, hepatocellular carcinoma or have refractory pruritus
- Liver transplantation improves cholestasis in 75 - 100% of patients, irrespective of PFIC subtype over a short term follow up of 3 - 5 years (Clin Res Hepatol Gastroenterol 2012;36:S26)
Microscopic (histologic) description
- PFIC1:
- Preserved lobular architecture and bland canalicular cholestasis devoid of inflammation
- Cholestasis, hepatocellular ballooning, acinar pseudorosettes
- Hepatocytes are orderly and small in early stage
- Giant cell transformation infrequent and focal
- Interlobular bile duct may be hypoplastic with paucity
- Fibrosis not characteristic in early biopsies
- Progression to micronodular cirrhosis by second to third decades of life (Semin Liver Dis 2011;31:3)
- PFIC2:
- Morphological heterogeneity
- Deranged lobular architecture
- Zone 3 predominant canalicular and hepatocellular cholestasis with khaki colored bile
- Neonatal hepatitis: giant cell transformation, lobular inflammation, hepatocyte necrosis, apoptosis and hepatocyte ballooning with bile ductular reaction - in early biopsies
- Centrilobular fibrosis progressing to bridging fibrosis and eventually to micronodular cirrhosis commonly seen within first decade of life (Semin Liver Dis 2011;31:3)
- PFIC3:
- Diffuse hepatocellular cholestasis with occasional canalicular and ductular cholestasis
- Portal fibrosis, expansion and florid ductular proliferation without bile epithelial injury, closely resembling extrahepatic biliary obstruction
- Extensive portal fibrosis progresses to biliary cirrhosis in advanced stages (Semin Liver Dis 2011;31:3, Orphanet J Rare Dis 2009;4:1)
- PFIC4, 5 and 6:
- Intralobular cholestasis with ductular reaction; hepatocellular ballooning; giant cell transformation and fibrosis with progression to micronodular cirrhosis (World J Gastroenterol 2020;26:7470)
Microscopic (histologic) images
Contributed by Puja Sakhuja, M.D., Surbhi Goyal, M.D. and Lavleen Singh, M.D.
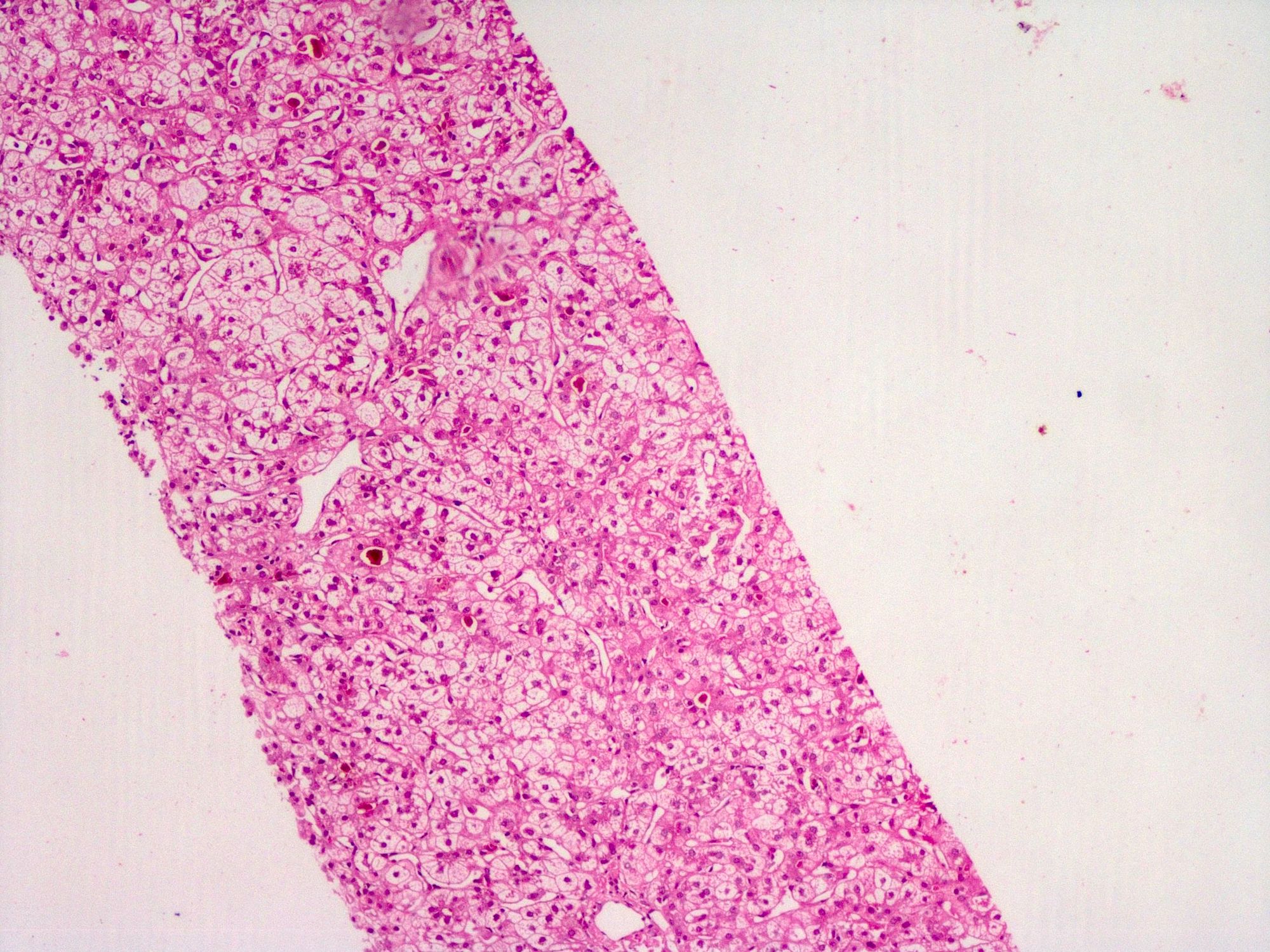
Bland cholestasis with hepatocytic pseudorosettes
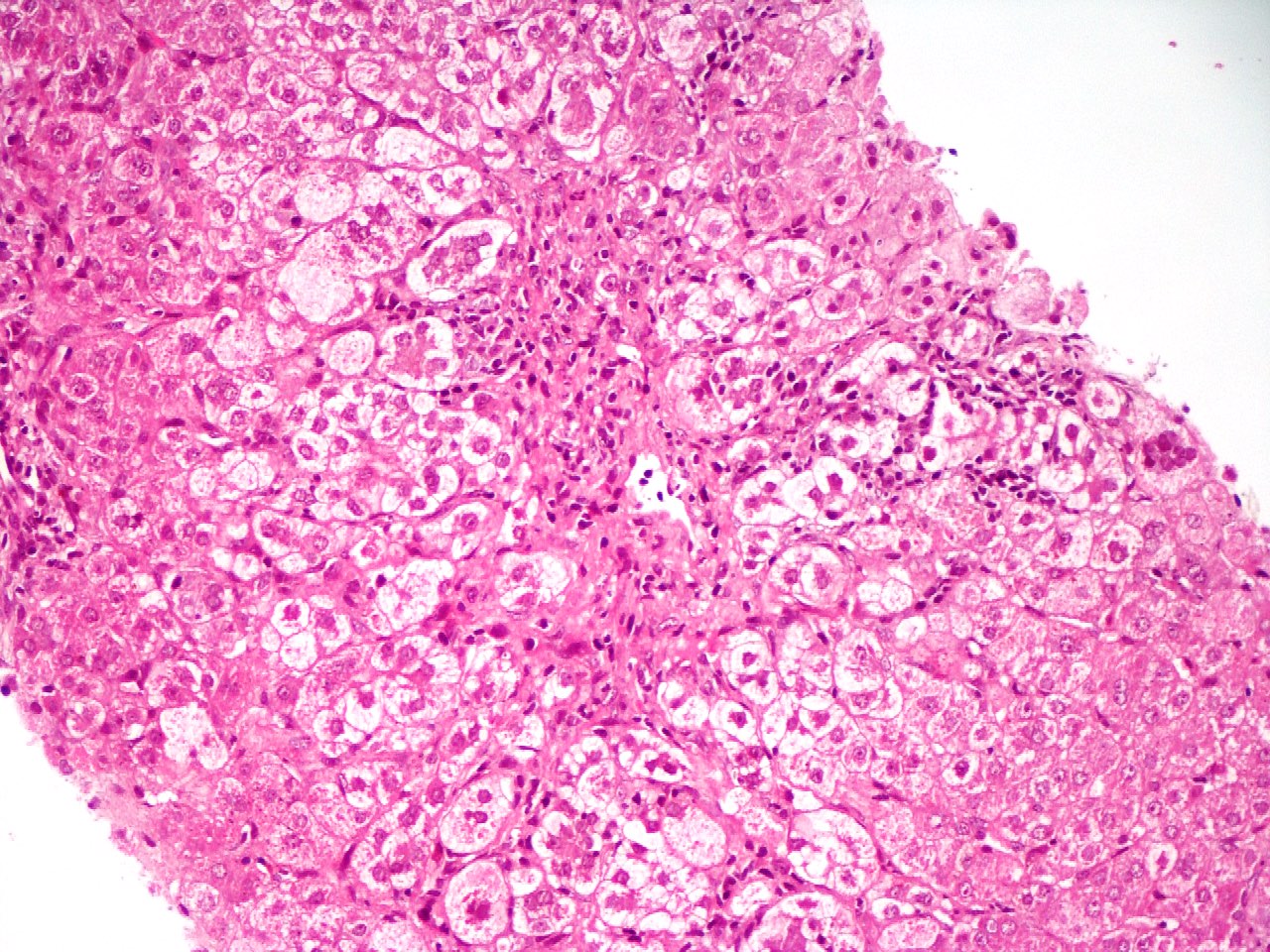
Giant cell
transformation
with pseudoacinar
rosettes
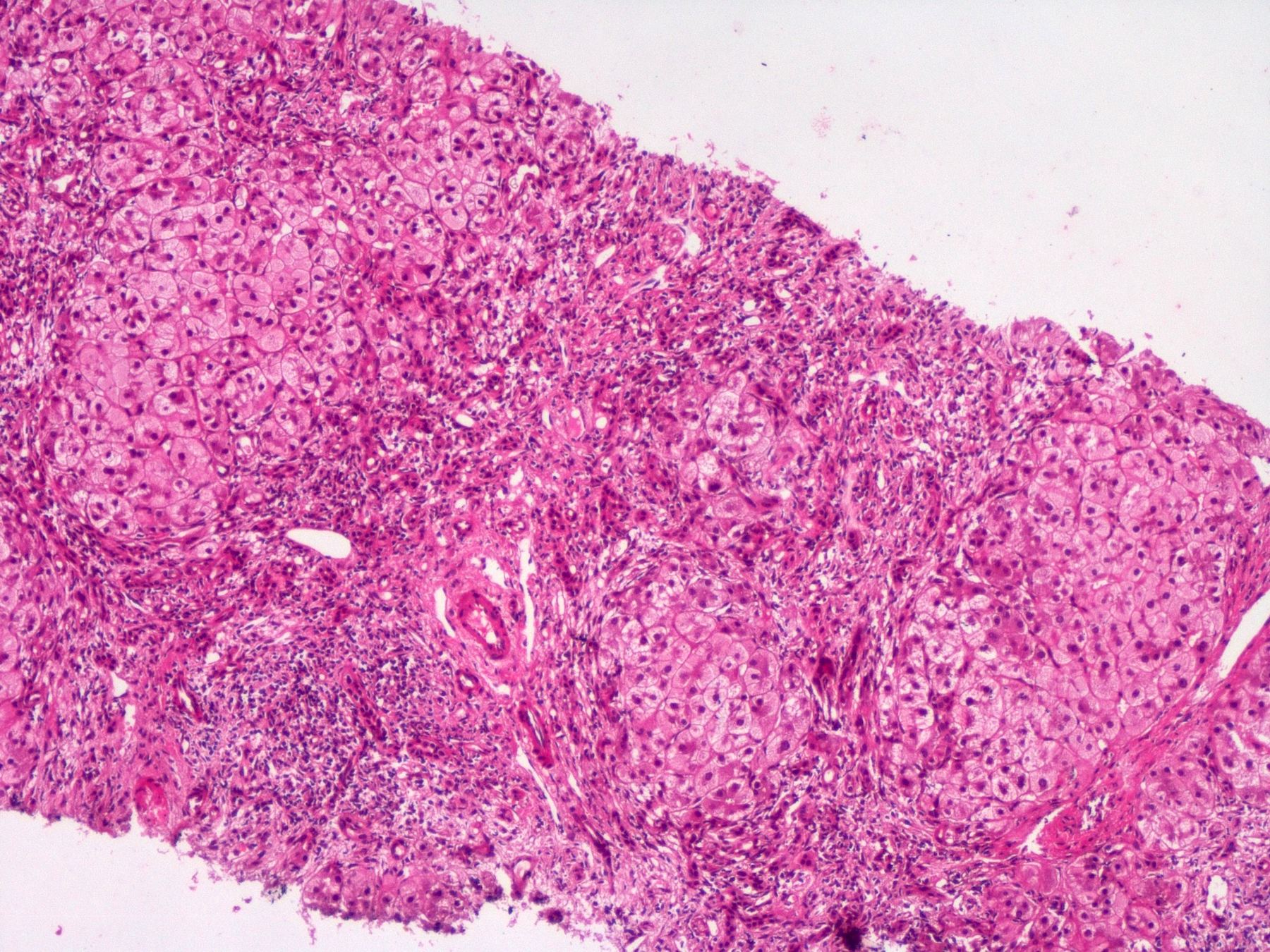
Extensive portal fibrosis with ductular proliferation
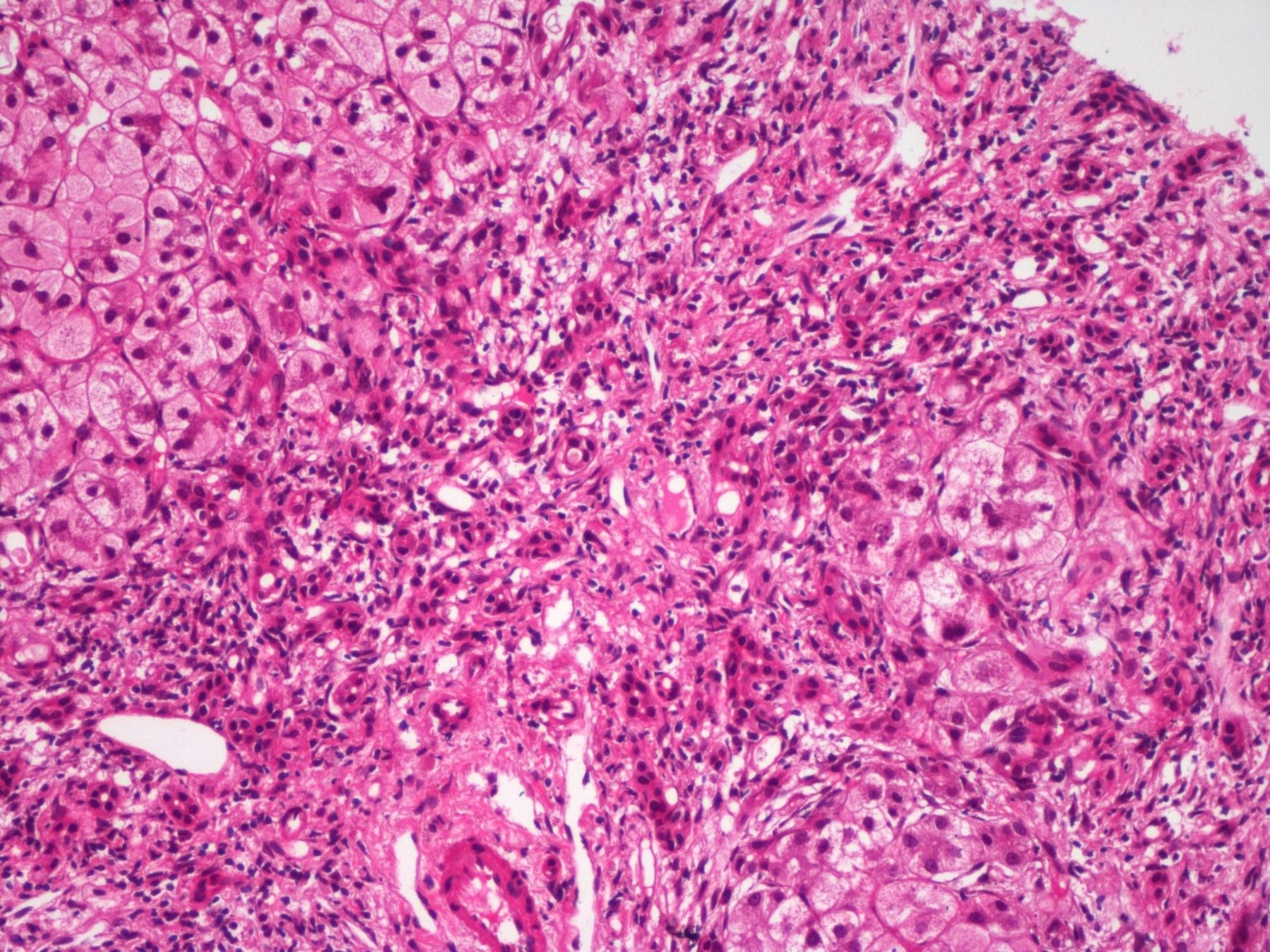
Cholestasis with ductular proliferation

Reduced canalicular expression of CD10
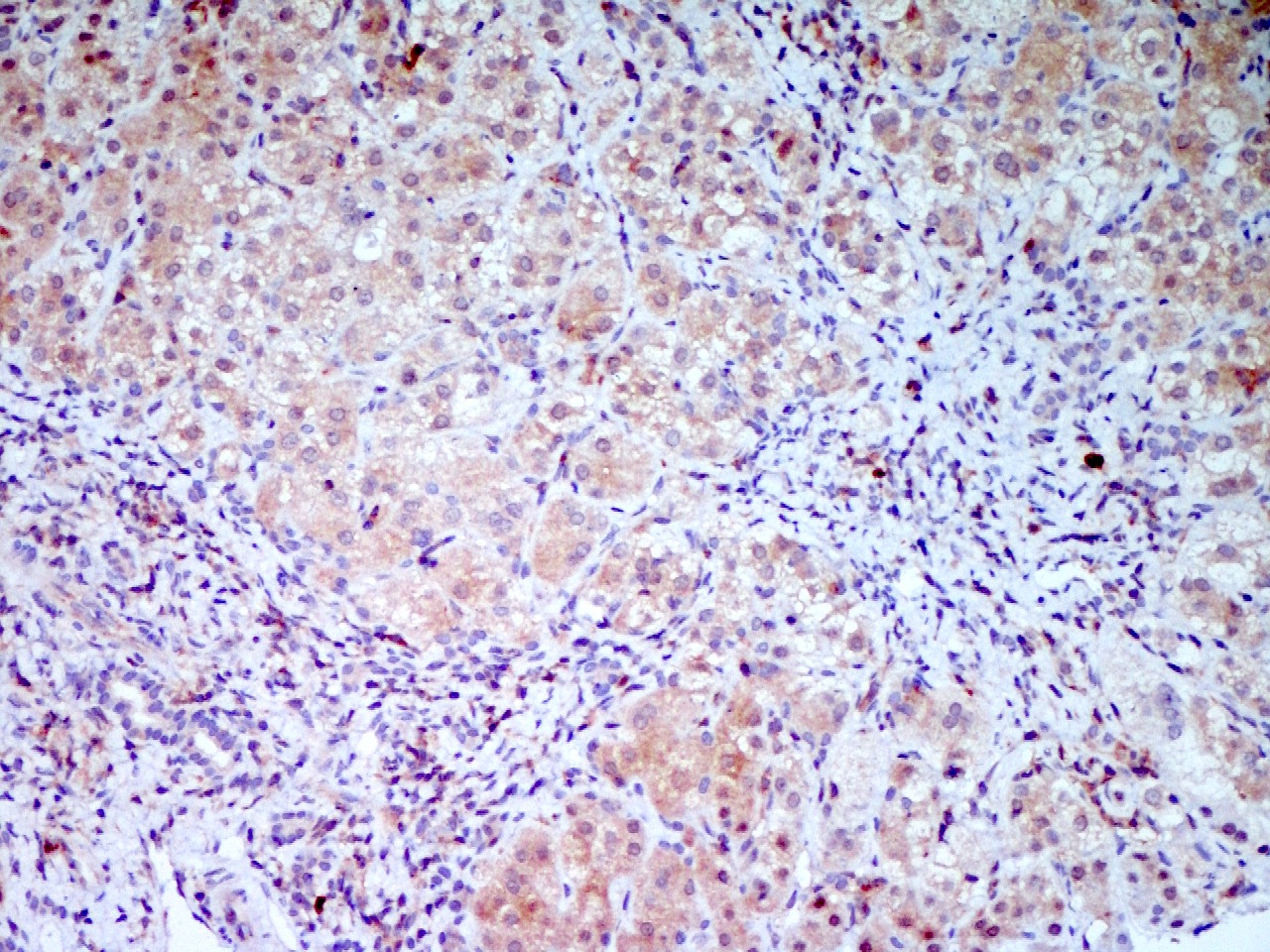
Loss of BSEP immunoexpression
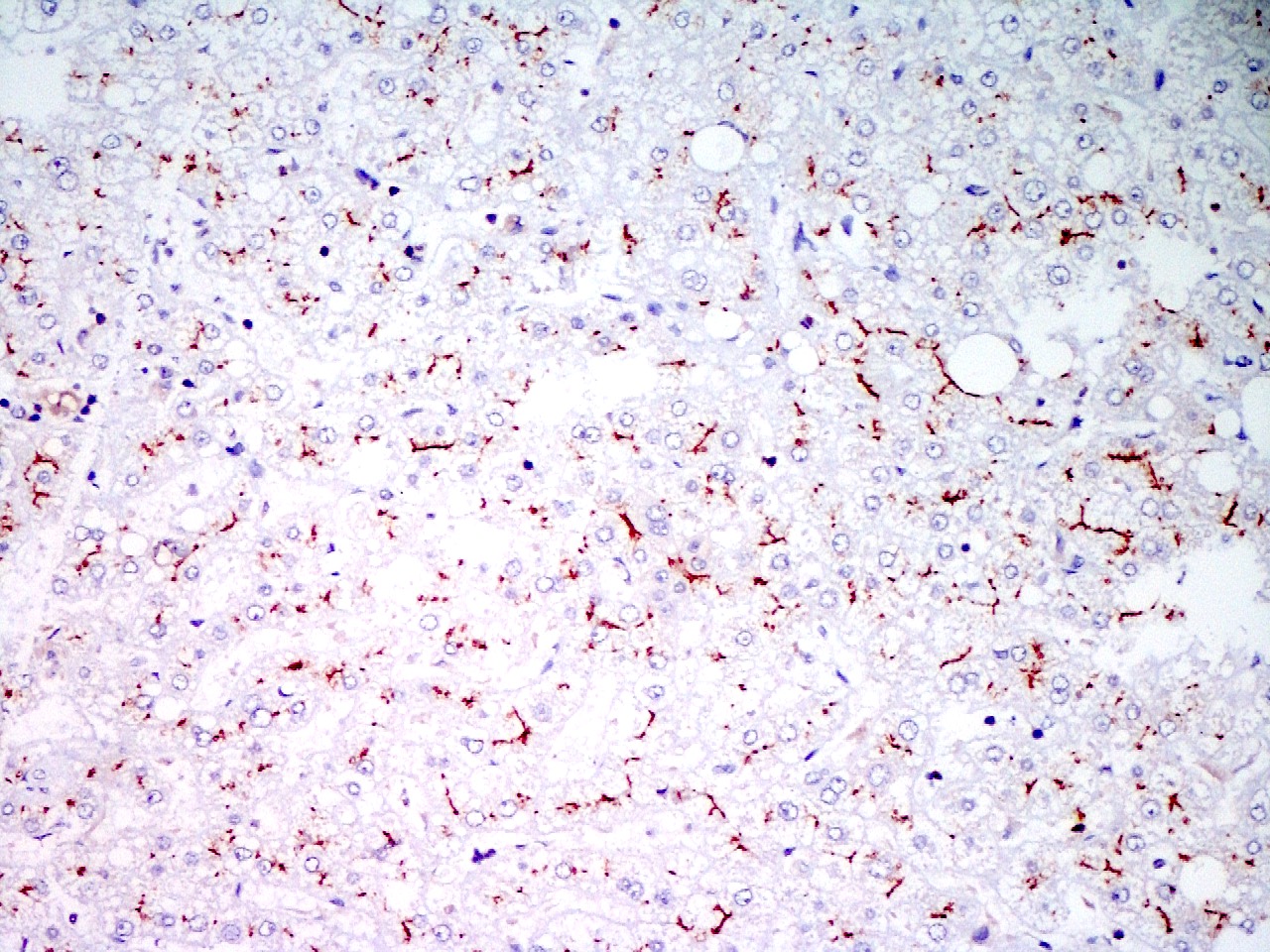
Canalicular BSEP expression
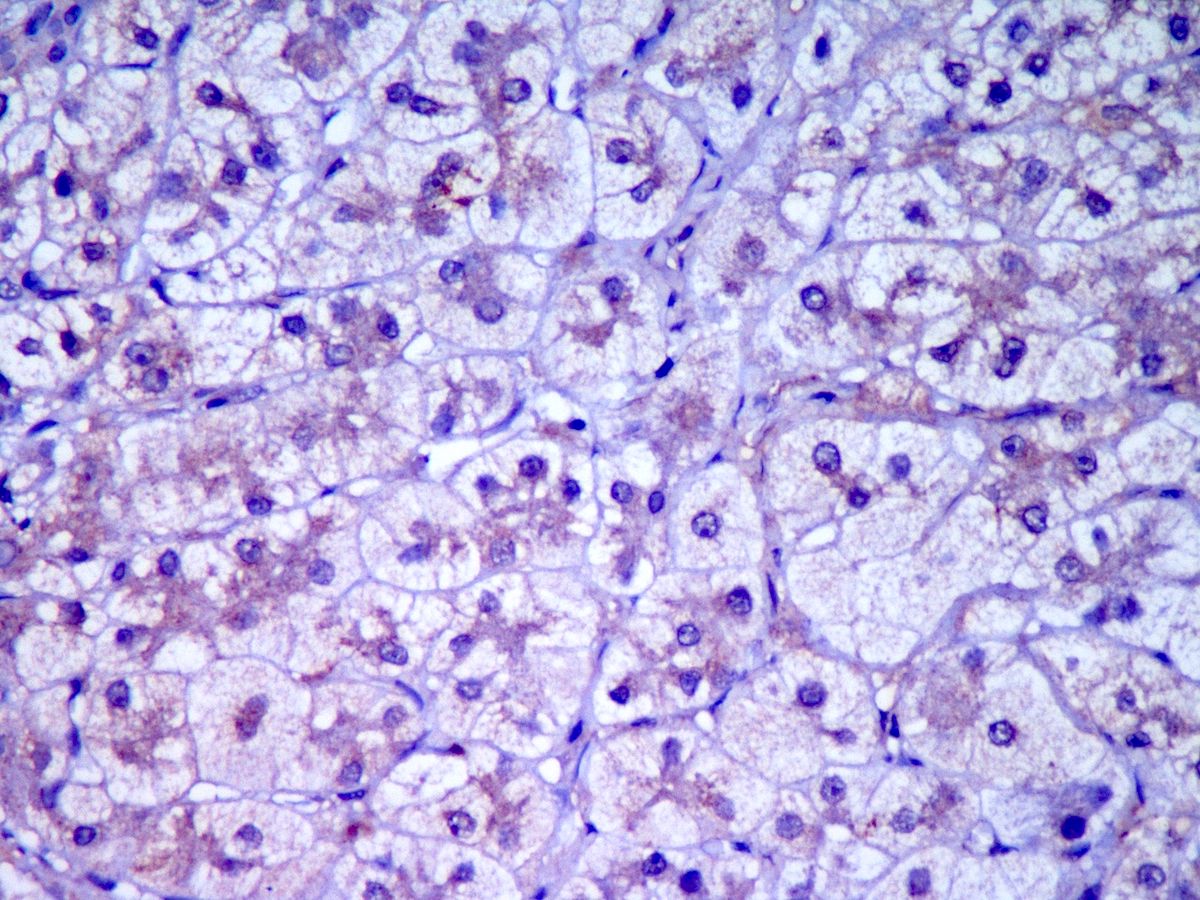
Loss of MDR3 immunoexpression
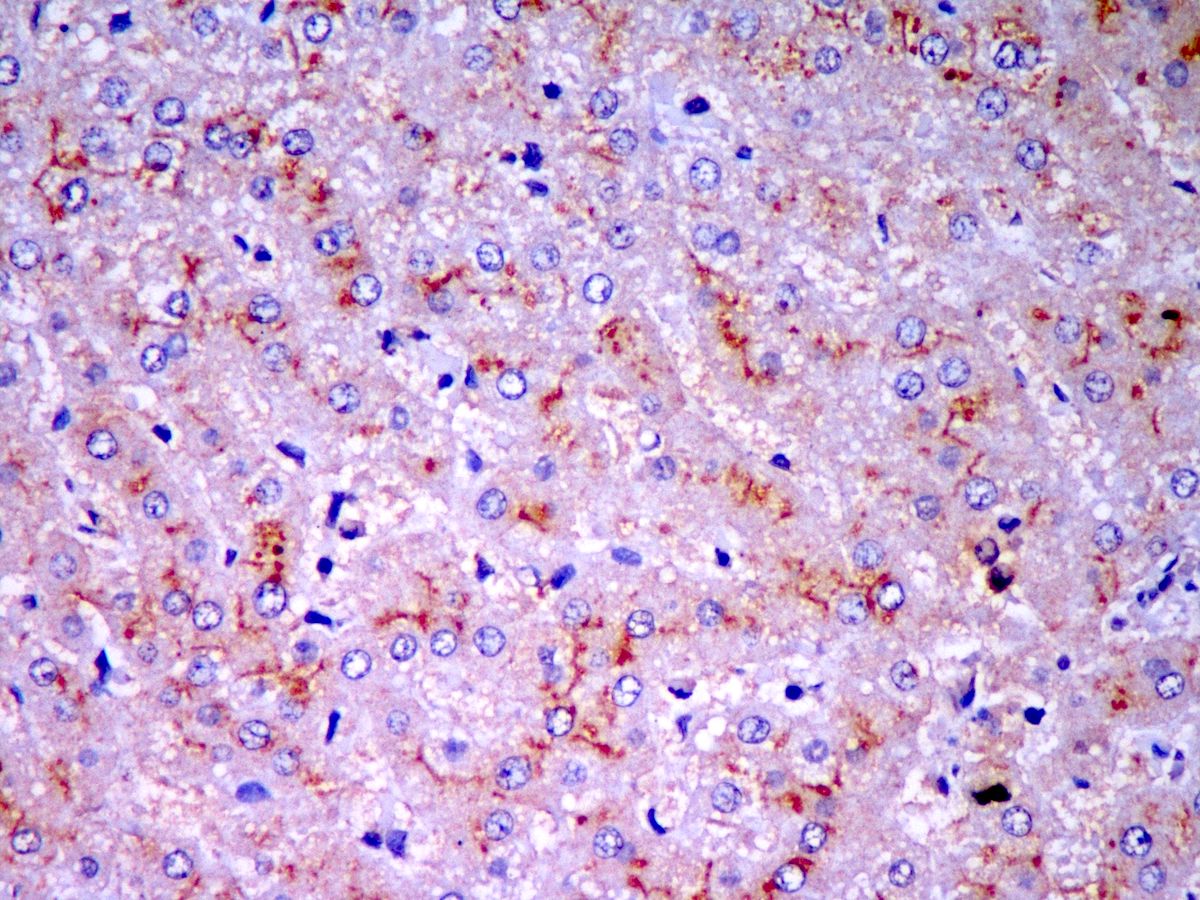
Canalicular MDR3 expression
Immunohistochemistry & special stains
- Antibody against ATP8B1 protein not validated yet
- PFIC1: canalicular markers GGT, CD10 and polyclonal CEA show a diminished or absent expression
- PFIC2 and 5: lack of canalicular expression of BSEP protein due to protein truncating mutations
- PFIC3: absent MDR3 protein expression (J Clin Exp Hepatol 2014;4:25)
- Weak / normal expression of BSEP and MDR3 can be seen in missense mutations; does not rule out functional deficiency (Orphanet J Rare Dis 2009;4:1)
- PFIC4: loss of TJP expression
- PFIC6: MYO5B and RAB11A show reduced canalicular staining and cytoplasmic expression, BSEP and MDR3 show patchy subcanalicular or absent expression (Clin Liver Dis 2018;22:657)
Electron microscopy description
- PFIC1: characteristic coarse granular Byler bile (J Pediatr 1972;81:484)
- PFIC2: amorphous fine filamentous bile
- PFIC3: presence of cholesterol crystals with angulated cleft-like lucencies (J Clin Exp Hepatol 2014;4:25)
Electron microscopy images
Contributed by Puja Sakhuja, M.D.
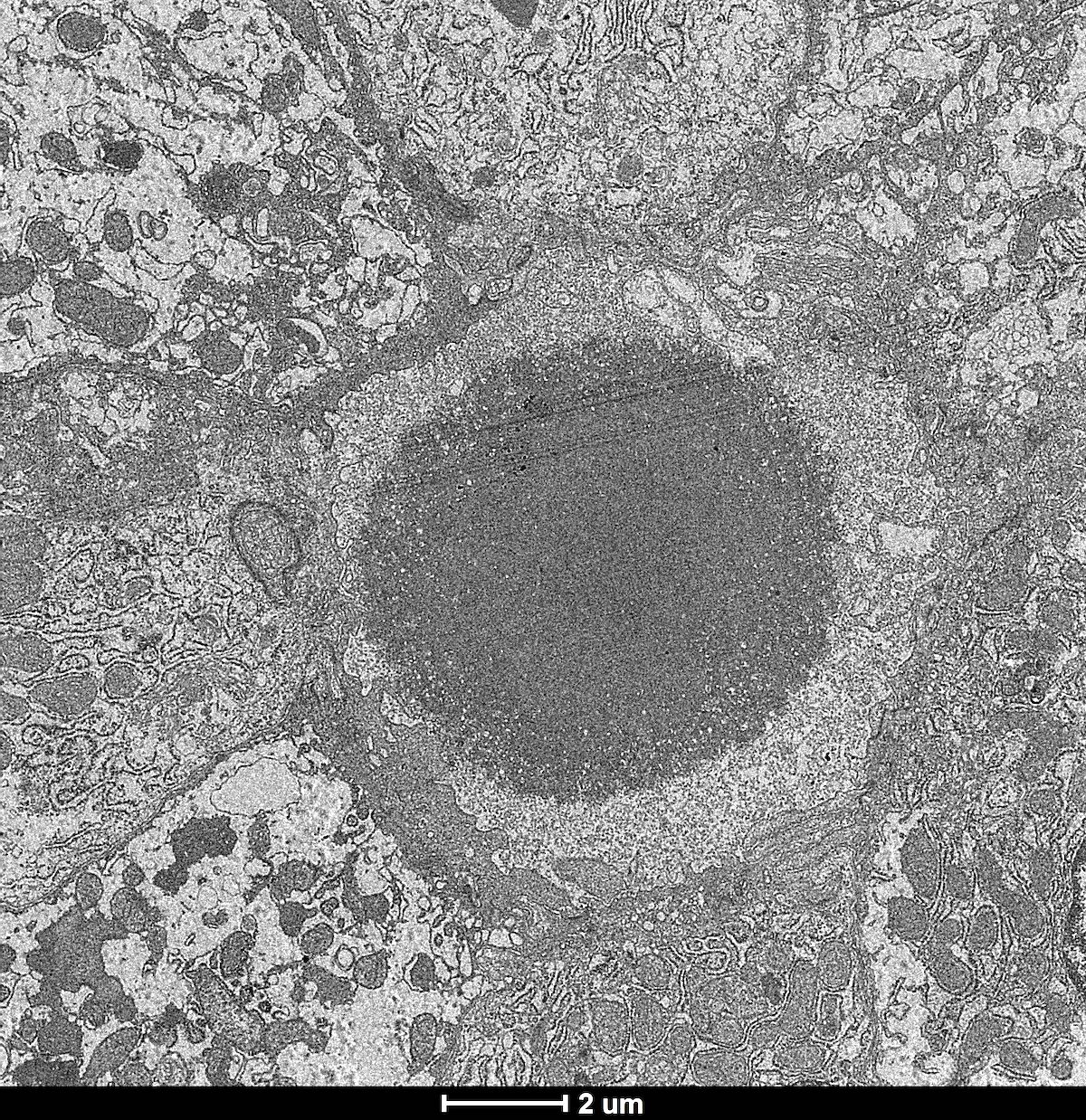
Amorphous, fine filamentous bile
Images hosted on other servers:

Distended bile canaliculi with coarse and granular bile
Molecular / cytogenetics description
- Autosomal recessive
- Detection of biallelic mutations is diagnostic
- PFIC1:
- More than 80 mutations in ATP8B1 gene on chromosome 18 (18q21)
- G308V mutation in the Amish population and D554N in the Greenland Inuits (Hepatology 2009;50:1597)
- PFIC2:
- More than 100 mutations in the ABCB11 gene on chromosome 2q24 - 31, which encodes the bile salt export pump (BSEP)
- E297G / D482G mutations in ABCB11 gene found in 30% of Europeans (J Hepatol 2010;53:170)
- PFIC3:
- Mutation in the ABCB4 gene, which encodes multidrug resistance protein 3, MDR3 (Orphanet J Rare Dis 2009;4:1)
- PFIC4:
- Mutations of the gene TJP2, encoding a tight junction protein
- PFIC5:
- Loss of function mutations in the NR1H4 gene (chromosome 12q23), encoding farnesoid X receptor (FXR), a bile acid activated, nuclear hormone receptor that regulates bile acid metabolism (Nat Commun 2016;7:10713)
- PFIC6:
- Missense or frameshift mutations in MYO5B, encoding the motor protein myosin Vb (myoVb) that facilitates protein trafficking in canalicular membrane (Hepatology 2014;60:301)
Sample pathology report
- Liver, biopsy:
- Neonatal hepatitis pattern of injury with loss of canalicular staining with BSEP on immunohistochemistry (see comment)
- Comment: The overall histologic picture, immunohistochemistry and electron microscopy findings support a diagnosis of PFIC type 2.
Differential diagnosis
- Extrahepatic biliary atresia:
- Presents as neonatal cholestasis and mimics PFIC3 on histology
- Ultrasound: absence of the gallbladder and the triangular cord sign (J Pediatr Surg 1999;34:1706)
- Cholangiography is confirmatory
- BSEP and MDR3 show normal canalicular expression; GGT is elevated
- Neonatal cholestasis / hepatitis:
- Exclude TORCH (toxoplasmosis, other, rubella, cytomegalovirus, herpes) and other viral infections by serological tests
- Metabolic screening, including urinalysis for reducing substances, ketones, copper excretion and gas chromatography mass spectrometry for organic acids, blood sugar, serum lactate, ammonia, alpha fetoprotein and ceruloplasmin levels
- GGT levels elevated
- Late infancy with pruritus:
- Bile acid synthetic defects:
- Present with cholestasis and normal GGT
- Serum bile acid concentration is low or absent
- Pruritus is mild or absent
- Response to bile acid therapy is good
- Drug induced cholestasis:
- Antimicrobials or acetaminophen may impair bile flow through hepatocellular damage, resulting in episodic cholestasis
- Arthrogryposis renal dysfunction syndrome (ARC syndrome) and familial Amish hypercholanemia:
- Rare cholestatic disorders with normal GGT levels
Additional references
Board review style question #1

A 1 year old boy presented with severe jaundice, watery diarrhea, failure to thrive and intractable pruritus. The investigations revealed normal liver transaminases, raised alkaline phosphatase and normal GGT, normal alpha-1 antityrpsin protein levels, reduced serum albumin and raised serum bile acids. Liver biopsy histology is shown above. Immunohistochemistry for BSEP is retained.
Which of the following is true regarding this entity?
- Diarrhea can get worse after liver transplantation
- Differential diagnosis includes serum bile acid synthetic defects in this case
- Histology shows lobular disarray with prominent giant cell transformation of hepatocytes and inflammation
- There is a risk of developing hepatocellular carcinoma or cholangiocarcinoma later in life
Board review style answer #1
A. Diarrhea can get worse after liver transplantation. Based on the history, investigations, histology and normal BSEP expression, the diagnosis in the above case is progressive familial intrahepatic cholestasis type 1 (PFIC1). Extrahepatic manifestations, like diarrhea, liver steatosis and short stature, do not improve and may even worsen after liver transplantation in PFIC1.
Comment Here
Reference: Progressive familial intrahepatic cholestasis
Comment Here
Reference: Progressive familial intrahepatic cholestasis
Board review style question #2
Which of the following is true regarding progressive familial intrahepatic cholestasis type 3 (PFIC3)?
- Bile acid analysis shows low bile acids with normal phospholipids
- Early onset vitamin K independent coagulopathy and markedly elevated AFP levels are characteristic of this entity
- Gamma glutamyl transferase levels are normal or low
- Normal immunoexpression of MDR3 does not exclude the diagnosis of PFIC3
Board review style answer #2
D. Normal immunostaining of MDR3 does not preclude the diagnosis of PFIC, as some missense mutations are associated with only functional defect in protein.
Comment Here
Reference: Progressive familial intrahepatic cholestasis
Comment Here
Reference: Progressive familial intrahepatic cholestasis
