

Thyroid & parathyroid
Hyperplasia / goiter
Dyshormonogenetic goiter
Copyright: 2003-2024, PathologyOutlines.com, Inc.
PubMed Search: Thyroid gland dyshormonogenetic goiter
See also: Aplasia / hypoplasia, Congenital hypothyroidism, Screening - neonatal hypothyroidism



- Thyroid enlargement due to various inherited defects in thyroid hormone synthesis
- Lack of circulating thyroid hormone activates TSH secretion, which causes overstimulation and hyperplasia of defective thyroid gland
- Important cause of congenital hypothyroidism often found during neonatal screening or otherwise manifested around puberty
- Sporadic goitrous cretinism was known before 20th century (for example, documented by Pendred and Osler), however biochemical defects and causative mutations of different entities began to be identified only after 1950 and 1990, respectively (Thyroid 2003;13:771)
- Less than 200 cases with detailed morphology have been reported (series and case reports), including around 30 cases with cancer
- Genetic autosomal recessive enzymatic defects in one of the steps of thyroid hormone synthesis, which causes congenital hypothyroidism (decreased to absent T3 / T4 and high TSH)
- Deficiency of thyroid peroxidase (TPO) is the most common cause worldwide
- Diffuse asymmetric enlargement of thyroid gland with prominent nodularity
- Histologic hallmarks are markedly hypercellular nodules with papillary hyperplasia, absence of colloid, frequent internodular bizarre cells and bridging fibrosis
- Dyshormonogenetic goiter = thyroid dyshormonogenesis
- Also known as inherited congenital / familial goiter and congenital / sporadic goitrous cretinism (more vague terms)
- Rare inherited disorder with incidence of 1 per 30 - 50K live births in Europe and North America
- Higher rates in consanguineous communities (Clin Endocrinol (Oxf) 2013;79:275)
- Second most common cause of congenital hypothyroidism (accounts for 10% - 15%) after thyroid dysgenesis
- F > M (2:1)
- Mean age at surgery is 16 years (range varies from neonates to sixth decade), with 80% cases occur before 25 years
- No racial or ethnic predilection (Thyroid 2011;21:13)
- Diffuse involvement of both lobes
- Mutations in genes encoding enzymes, which participate in thyroid hormone synthesis
- There are 7 genes involved in thyroid hormonogenesis and all related steps may be impaired: thyroglobulin synthesis (TG), iodide transport via basal (NIS/SLC5A5) and apical (PDS/SLC26A4) membranes of follicular cell, generation of H2O2 (DUOX2, DUOXA2), iodide organification (TPO), coupling of mono- and diiodotyrosine (TPO), proteolytic breakdown of thyroglobulin and iodide recycling (IYD/DEHAL1)
- TPO (OMIM 274500) and TG (OMIM 274700) are the most frequent responsible genes (Thyroid Res 2015;8:5)
- Mostly due to single gene mutation, multiple mutations is more uncommon
- The earlier the enzyme abnormality occurs in pathway of hormone synthesis, the more severe the clinical presentation (Endocr Rev 1983;4:213)
- Mode of inheritance of all forms of thyroid dyshormonogenesis is autosomal recessive, with the exception of DUOX2 mutations, which may be autosomal dominant (Endocr Dev 2014;26:60)
- Mechanism: impaired T3 / T4 synthesis → deficient circulating thyroid hormone → loss of negative feedback loop → normal physiologic response to increased thyrotropin releasing hormone production in hypothalamus → increased TSH secretion in anterior pituitary → chronic TSH stimulation → hyperplasia of thyroid gland
Images hosted on other servers:
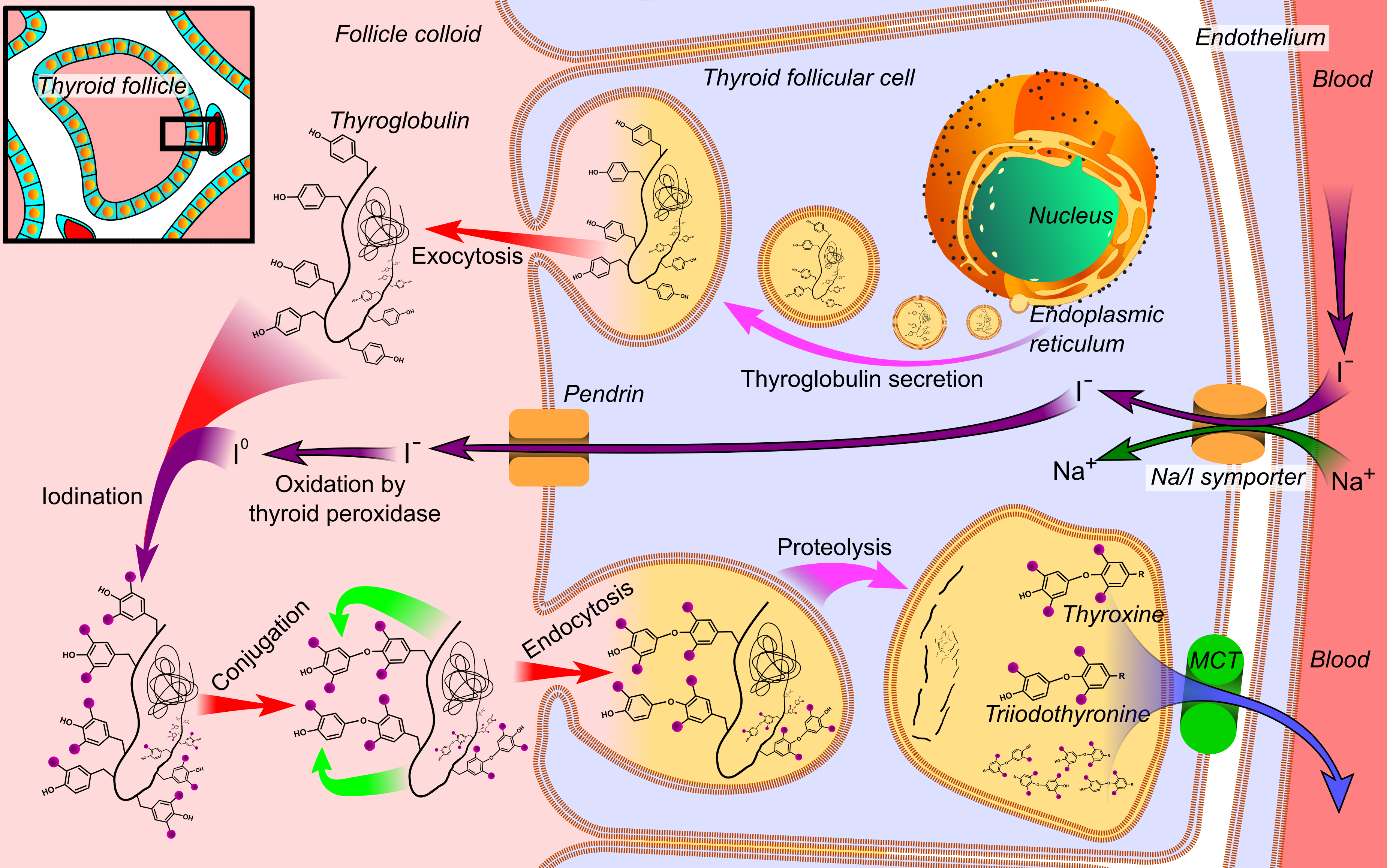


Thyroid hormone synthesis

Mechanism of
dyshormonogenesis
Congenital hypothyroidism

Genetic causes of
dyshormonogenesis

Thyroid imaging in congenital hypothyroidism

Scintigraphic classification of dyshormonogenesis




Pedigrees
- Various degrees of hypothyroidism and goiter are seen
- Clinical presentation depends on severity of the metabolic error (Wenig: Atlas of Head and Neck Pathology, 3rd Edition, 2015)
- Early presentation in neonatal period with evident goiter and congenital hypothyroidism complicated by growth and intellectual disability (cretinism) suggests severe defect
- Presentation in puberty or adult life with goiter and minimal evidence of thyroid dysfunction (euthyroidism, subclinical hypothyroidism) implies mild defect
- Some metabolic defects may be masked by a high dietary iodine intake, evident from Japanese series (Endocr Dev 2014;26:60)
- Familial recurrence of congenital hypothyroidism due to dyshormonogenesis is 25% (J Clin Endocrinol Metab 2014 Feb;99:363)
- Most cases manifest before third decade
- Very rarely a diagnosis is made only at autopsy
- Pendred syndrome (Pendred Syndrome/DFNB4, J Med Genet 1996 Dec;33:1037)
- Familial association of congenital goiter with deaf mutism (most patients presents with severe deafness rather than with hypothyroidism)
- Caused by mutations (150+ variants) in PDS / SLC26A4 gene encoding pendrin (OMIM 274600)
- Occurs in 7.5 to 20 per 100,000 individuals
- Accounts for 10% of patients with hereditary deafness
- With sufficient dietary iodide, 90% are clinically and biochemically euthyroid (Curr Opin Pediatr 2011 Aug;23:421)
- No associated malformations are present in dyshormonogenetic goiter except Pendred syndrome (J Clin Endocrinol Metab 2014 Feb;99:363)
- Tumors may develop in dyshormonogenetic goiter (see Case Reports)
- Most common are follicular carcinoma and incidental papillary microcarcinoma
- Also papillary carcinoma and rarely follicular adenoma
- Dyshormonogenesis may predispose to follicular carcinoma or follicular variant PTC by increasing chance of mutation in dividing follicular cells under chronic TSH stimulation (Clin Endocrinol Metab 1979;8:193) but additional genetic hits may be needed, such as p53 mutation (Endocr Pathol 2016;27:70)
- History of congenital or familial goiter in a young individual (often diagnosed due to routine screening of newborns for congenital hypothyroidism)
- Radiologic evaluation includes thyroid ultrasound and scintiscan
- Correlation of clinical signs with laboratory and ancillary tests
- Laboratory workup is complex - goal is identifying impaired step of thyroid hormone synthesis
- Prenatal diagnosis can be made in utero by sonography and measurement of TSH in amniotic fluid (J Obstet Gynaecol Res 2013;39:720)
- Hypothyroidism (if present): elevated TSH, low serum T4, T3 and thyroglobulin
- Additional tests (J Med Genet 2005;42:379): thyroxine binding globulin, thyrotropin releasing hormone (to evaluate hypothalamic-pituitary axis), thyroglobulin antibodies (to rule out autoimmune thyroiditis), urinary mono and diiodotyrosine, oral radioiodine test (to measure iodide trapping via saliva to plasma ratio)
- Ultrasound reveals enlarged, orthotropic thyroid (Korean J Radiol 2015;16:419)
- Solid hypoechoic nodules
- Lobes develop a convex appearance laterally, and isthmus is readily visualized, in contrast to a normal gland
- Thyroid gland size in children varies with age and should be correlated with height, weight and body surface area
- Ultrasound scans at 20 to 22 weeks gestation are able to detect fetal thyroid hypertrophy (J Clin Endocrinol Metab 2014;99:363)
- Scintigraphy (Endocr Dev 2014;26:60, Pediatr Radiol 2013;43:1244)
- In iodide trapping defect (NIS), patients show no or very low radioiodine uptake in the thyroid; further, in contrast to athyreosis, salivary glands and the gastric mucosa lack uptake ("white scintigraphy")
- In iodide organification defect (TPO, DUOX2, PDS), radioiodine uptake is normal, but perchlorate discharge test is positive - most radioisotope is washed out into the bloodstream instead of accumulating in thyroid
- Patients with TG and DEHAL1 defects show normal radioiodine uptake and normal perchlorate test
Images hosted on other servers:
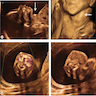
Ultrasound of fetal goiter
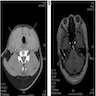
Ultrasound of 26 year old man
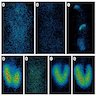
Various defects
on scintigraphy
(NIS, TPO and PDS)
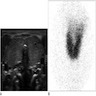
Scintigraphy
- Boy with thyroid peroxidase gene defect (Clin Endocrinol (Oxf) 2006;64:514)
- 5 and 10 year old brothers with congenital goiter due to TPO mutation (J Pediatr Endocrinol Metab 2016;29:627)
- 7 year old girl with DUOXA2 mutation (J Clin Endocrinol Metab 2008;93:605)
- 22 year old woman with Pendred syndrome (J Pediatr Endocrinol Metab 2008;21:1179)
- 35 year old man with Pendred syndrome and retrosternal goiter (Indian J Surg 2013;75:329)
- 38 year old man with iodide transport defect (Endocr Pathol 1998;9:225)
- Case with thyroglobulin synthesis defect (Thyroid 1996;6:11)
- Two families with TPO mutations (Exp Clin Endocrinol Diabetes 2013;121:343)
- Dyshormonogenetic goiter with clear cell change resembling parathyroid adenoma (Indian J Pathol Microbiol 1998;41:469)
Cancers in dyshormonogenetic goiter
- Female infant with metastatic follicular thyroid carcinoma arising from congenital goiter due to TPO mutation (J Clin Endocrinol Metab 1998;83:4162)
- 6 year old girl with papillary carcinoma and congenital dyshormonogenetic hypothyroidism (Diagn Cytopathol 2009;37:707)
- 14 year old boy with follicular adenoma and papillary carcinoma arising in dyshormonogenetic goiter (Univ Pittsburgh Case #425)
- 17 year old girl with metastatic papillary carcinoma arising from dyshormonogenetic goiter (Case Rep Med 2013:813167)
- 26 year old woman with follicular thyroid carcinoma and dyshormonogenetic goiter due to TPO gene mutation (Thyroid 2012;22:542)
- 31 year old woman with metastatic papillary thyroid carcinoma in goiter caused by TG mutation (J Clin Endocrinol Metab 2010;95:1000)
- 35 year old man with papillary thyroid carcinoma arising from dyshormonogenetic goiter (Endocrinol Jpn 1987;34:955)
- 37 year old man with rare and particular form of goiter (Ann Transl Med 2013;1:21)
- 53 year old woman with Pendred syndrome and metastatic follicular thyroid carcinoma with anaplastic transformation (Thyroid 2001;11:981)
- 65 year old woman with Pendred syndrome associated thyroid carcinoma (Endocr Pathol 2016;27:70)
- 66 year old woman with hyperfunctioning metastatic follicular thyroid carcinoma in Pendred syndrome (Cancer 1991;67:2191)
- Two siblings with congenital goiter and metastatic follicular carcinoma (J Clin Endocrinol Metab 1981;52:294)
- T4 replacement therapy until the serum TSH becomes normalized
- Some disorders can be treated with iodine supplementation, e.g. all SLC26A4, DUOX2, DUOXA2 and IYD defects and partial defects in SLC5A5 (Curr Opin Pediatr 2011;23:421)
- Surgery is considered in patients with very large goiters (compressive or cosmetic reasons) or when malignancy is suspected
- Fetal goiter detected by ultrasound with confirmed hypothyroidism (high amniotic TSH) can be treated by intra-amniotic injections of thyroxine (J Obstet Gynaecol Res 2013;39:720)
Images hosted on other servers:
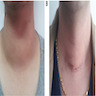
Pendred syndrome
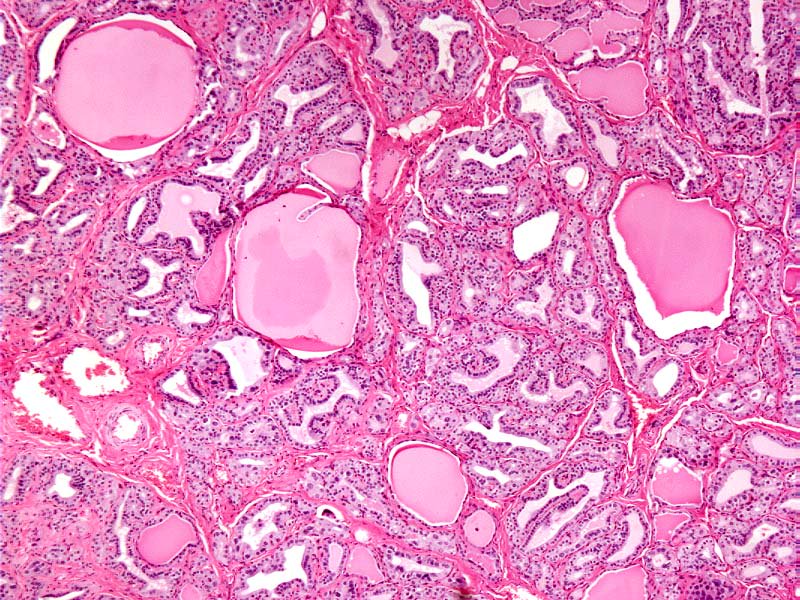
- Asymmetrically enlarged thyroid gland with multiple nodules, grossly indistinguishable from multinodular goiter
- Adult weight is 50 to 250 g, with exceptional cases over 2000 g (Endocr Pathol 1997;8:283, J Pathol 1969;99:251)
- Cut surface is firm and tan with pale nodules of varying size (1 to 6 cm), which may be encapsulated (J Pathol 1969;99:251)
- Internodular tissue is scant, delicately lobular and medullary to myxomatous, totally lacking the "beefy" texture of normal thyroid gland (Am J Pathol 1955;31:997)
- Secondary changes include old and fresh hemorrhage, fibrosis, cyst formation, rarely calcification (Endocrine Pathology 1994;5:49)
AFIP images

Marked nodular hyperplastic changes
Images hosted on other servers:



Enlarged multinodular thyroid
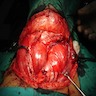
Excision

Follicular adenoma in a goiter
- Diffuse process with prominent nodularity and no normal appearing thyroid tissue (Wenig: Atlas of Head and Neck Pathology, 3rd Edition, 2015)
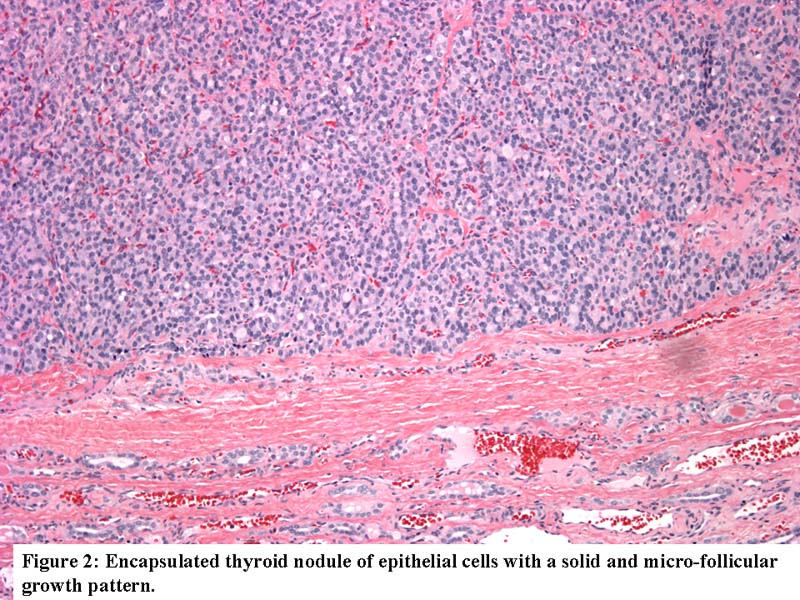
- Nodules and internodular thyroid parenchyma are markedly cellular/hyperplastic (Nikiforov: Diagnostic Pathology and Molecular Genetics of the Thyroid, 2nd Edition, 2012)
- Predominant pattern of nodules is solid or microfollicular, also trabecular, insular and macrofollicular / microcystic
- Papillary hyperplasia prominent within the nodules; papillae are mainly simple (without fibrovascular cores and branching) and broad
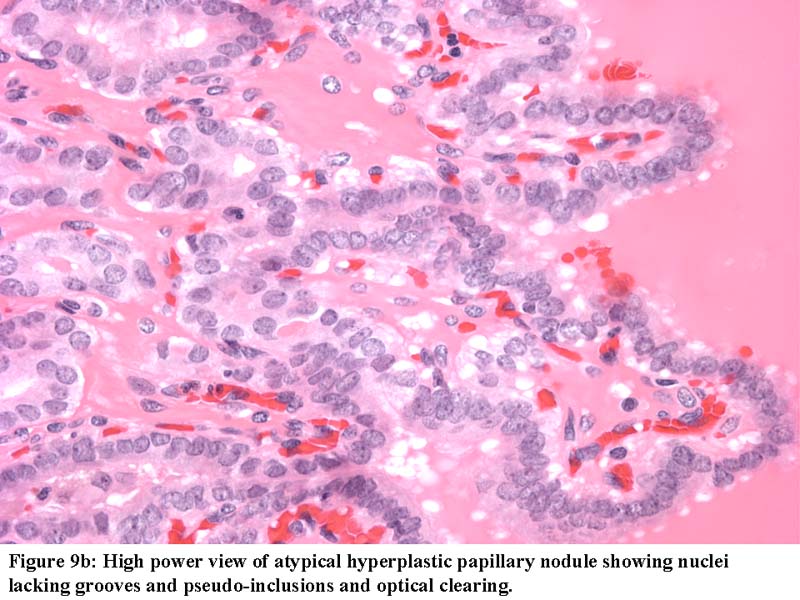
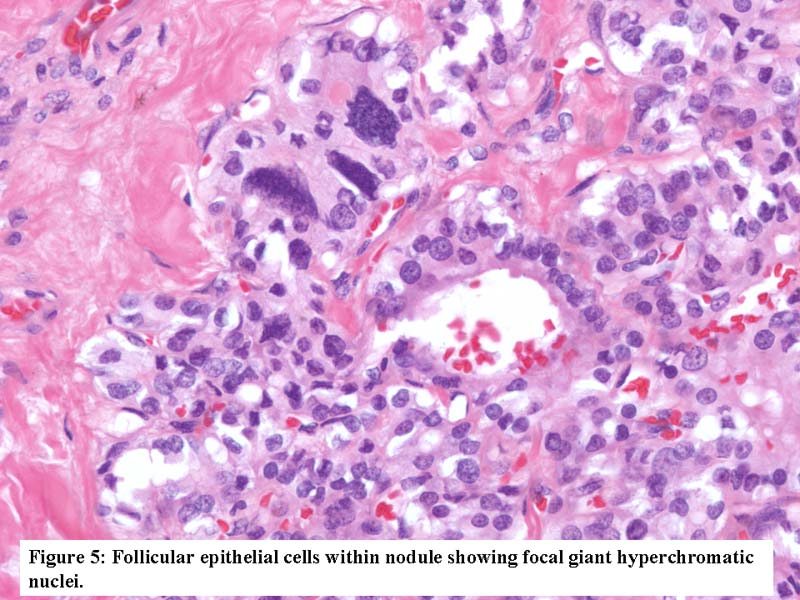
- Marked nuclear atypia with bizarre nuclei (enlarged, irregularly shaped, hyperchromatic), more evident in internodular tissue than in the nodules; also random vesicular nuclei, occasional mitotic figures
- No / minimal colloid ("empty follicles") within the entire gland; when present, it is usually pale, filamentous and found in apparent nodules
- "Kaleidoscopic" pattern of nodules is remarkable: different nodules show different degrees of cellularity and architecture (Am J Pathol 1955;31:997), and any one nodule is not uniform in structure (J Pathol 1969;99:251)
- Internodular background is uniform in structure
- Associated changes
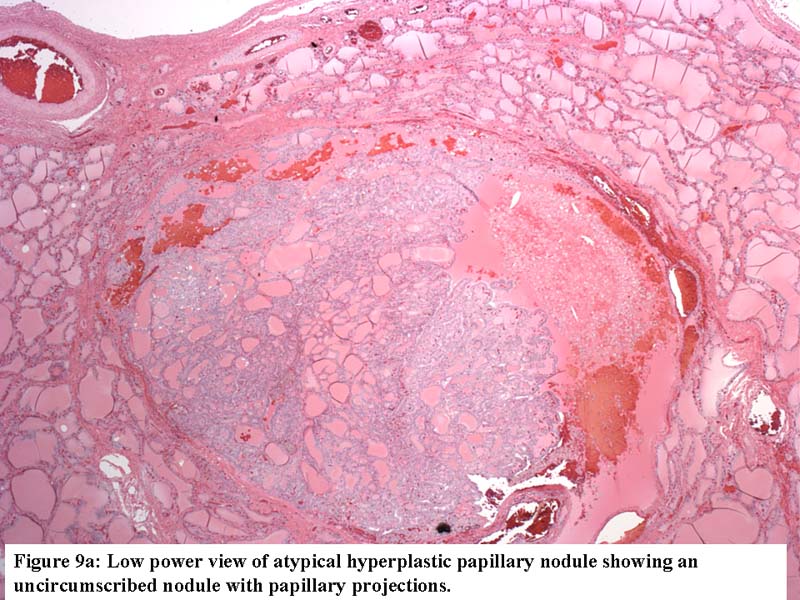
- Extensive bridging fibrosis of internodular tissue; sometimes may circumscribe nodules and cause irregularities at the border simulating capsular invasion
- Cytoplasmic oxyphilia (sign of functional exhaustion) and focal clear cell metaplasia (sign of hormone overproduction) (Int J Surg Pathol 1999;7:3)
- Myxoid change
- Absent or rare focal mild thyroiditis
- Important considerations
- Histologic features of the background thyroid are as important as the appearance of the nodules (Int J Surg Pathol 1999;7:3 )
- Histologic changes in newborns are not as prominent as in older patients, who develop variation of patterns and secondary changes over the course of disease (J Pathol 1969;99:251)
- Severity of microscopic appearance depends on degree and duration of TSH stimulation and is modulated by therapy (hormone replacement or iodine supplementation)
- Dyshormonogenetic goiter should be suspected in a child or young adult with diffuse follicular hyperplasia, nodularity, scant colloid and nuclear atypia
- Several studies (J Pathol 1969;99:251, Endocrine Pathology 1994;5:49) found that structural changes in the gland may correlate with particular defects (e.g. microcystic / alveolar pattern in cases with formation of abnormal iodoprotein, or severe atypia and solid pattern in organification defects), however most modern authors believe that morphology is independent of the biochemical abnormality (Mills: Sternberg's Diagnostic Surgical Pathology, 6th Edition, 2015, Wenig: Atlas of Head and Neck Pathology, 3rd Edition, 2015)
- Cancer in dyshormonogenetic goiter
- Nuclear and architectural features typical for dyshormonogenesis often mimic malignancy
- In one study almost every fifth case was considered as follicular, papillary, medullary, or undifferentiated carcinoma (Endocr Pathol 1997;8:283)
- Some previously reported cases of congenital thyroid carcinoma were actually examples of thyroid dyshormonogenesis (Clin Endocrinol Metab 1981;10:317)
- Only strict criteria for diagnosing malignancy should be applied, e.g. true capsular or vascular invasion in case of follicular carcinoma
Scroll to see all images:
Contributed by Mark R. Wick, M.D. and AFIP
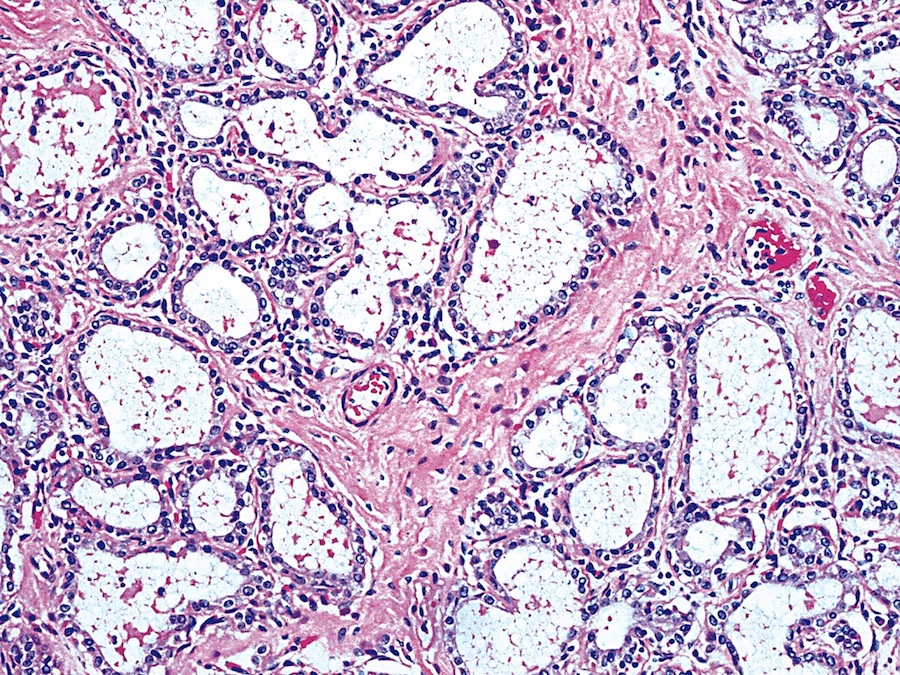

Various images
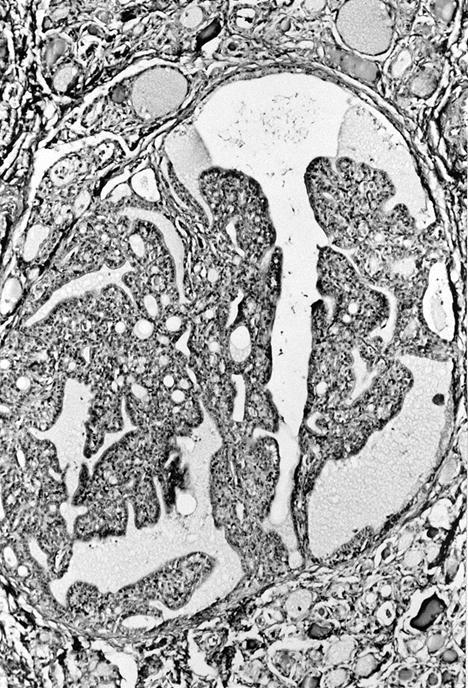
Resembles nodular hyperplasia
Hyperplastic nodules showing solid growth pattern

Marked fibrosis causes apparent increased lobularity
Images hosted on other servers:


Multinodularity

Fibrosis



Hypercellular nodule
Papillarity

Hyperplastic nodule


Scant colloid
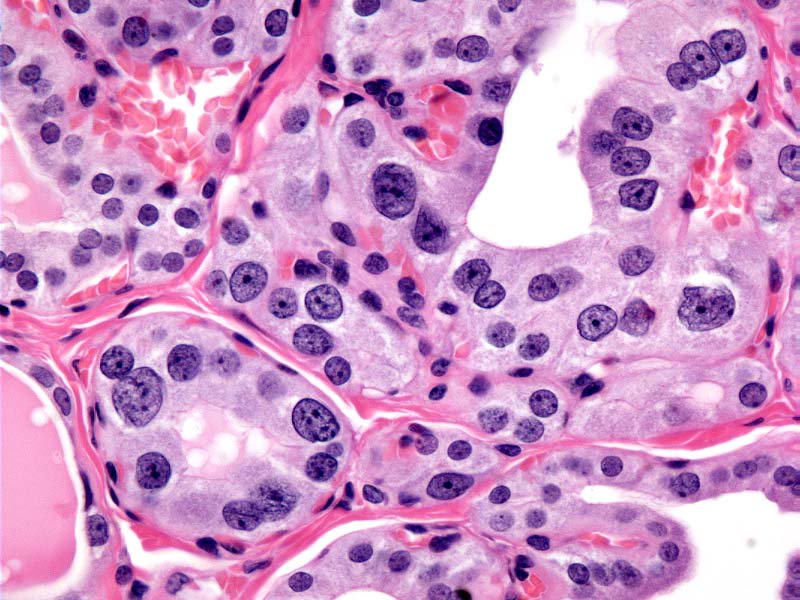
Atypia

IHC in Pendred syndrome


Follicular adenoma in dyshormonogenetic goiter
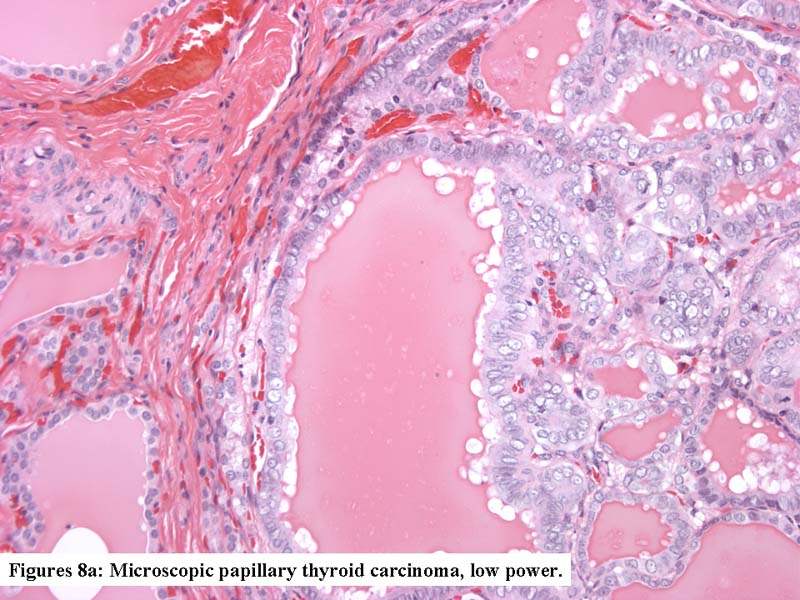
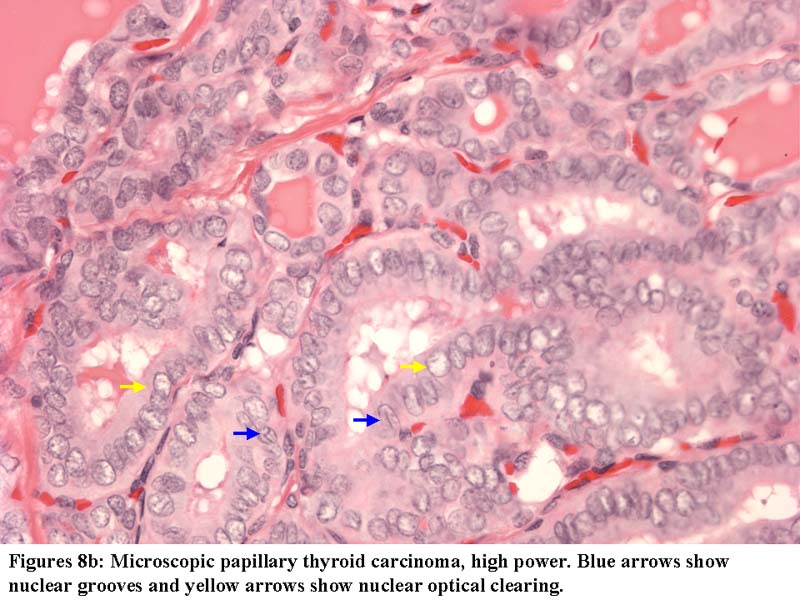
Papillary microcarcinoma in dyshormonogenetic goiter



Papillary carcinoma in goiter
- Highly cellular aspirate with syncytial fragments arranged in predominantly microfollicular pattern (Indian J Pathol Microbiol 2001;44:169, Diagn Cytopathol 2013;41:720)
- Follicular cells with round to oval nuclei, sometimes atypical or vesicular, but without grooves / inclusions
- No / scant colloid
- Fragments of fibrocollagenous tissue
- Marked hyperplastic changes closely resemble follicular lesion and follicular neoplasm (Diagn Cytopathol 2005;33:252)
- Cytology alone is not able to differentiate dyshormonogenesis from tumor
- Fine needle biopsy is not recommended for dyshormonogenetic goiter (Kini: Thyroid Cytopathology: An Atlas and Text, 2nd Edition, 2015)
- Rarely presents as benign cyst (Arq Bras Endocrinol Metabol 2014;58:958)
- Thyroglobulin and TTF1, but thyroglobulin may show a marked decrease in follicular lumina in dyshormonogenesis due to TG mutations (J Clin Invest 1996;98:2838, J Clin Endocrinol Metab 2010;95:1000)
- Weak staining with antibodies against corresponding targets of dyshormonogenesis
- Faint TPO immunostaining in cases with TPO defects (Rev Hosp Clin Fac Med Sao Paulo 1998;53:86)
- Weak staining with pendrin in Pendred syndrome (J Clin Endocrinol Metab 2008;93:267)
- Increased proliferation rate by Ki67 in solid nodules (Indian J Pathol Microbiol 2001 Apr;44:169)
- PAS can detect traces of colloid, if not evident on H&E (Wenig: Atlas of Head and Neck Pathology, 3rd Edition, 2015)
- No diffuse positivity for calcitonin and neuroendocrine markers (e.g., chromogranin, synaptophysin), however C cells are present within the gland (Endocrine Pathology 1994;5:49)
- Tall follicular cells with long and numerous microvilli reflecting high metabolic activity (Endocr Pathol 1997;8:283)
- Cytoplasm with numerous mitochondria and abundant rough endoplasmic reticulum with vesicular dilated cisternae (J Clin Invest 1996;98:2838)
- Early genetic screening is justified even without complete clinical workup, because a precise molecular diagnosis allows genetic counseling and the identification of asymptomatic mutation carriers at risk of recurrent hypothyroidism, and provides a rationale for treatment (Curr Opin Pediatr 2011;23:421, J Clin Endocrinol Metab 2014;99:363)
Images hosted on other servers:


Structure of TPO mutations

DUOXA2 mutation
- Multinodular endemic goiter (Endocrine Pathology 1994;5:49)
- Similar: identical gross appearance, extensive hyperplasia
- Difference: abundant colloid, hyperplasia confined by nodules (not throughout the entire gland), normal thyroid tissue present between nodules, frequent secondary degenerative changes (hemorrhage, foamy and hemosiderin laden macrophages, dystrophic calcification, fibrosis and cyst formation)
- Ancillary studies: euthyroidism, rarely toxic goiter
- Radiation thyroiditis: diffuse cytologic atypia through gland, history of thyroid radiation
- Iatrogenic goiter caused by antithyroid drugs: similar histology, history of thionamides treatment of patient or mother (in case of fetal goiter)
- Graves disease (especially untreated): diffuse hyperplasia without nodularity, no solid or related patterns, columnar epithelium present without nuclear atypia, hyperthyroidism clinically
- Follicular adenoma: unilocular lesion
- Follicular thyroid carcinoma with capsular invasion
- Localized lesion with otherwise uninvolved thyroid parenchyma
- Clear cut capsular or vascular invasion must be present; capsule is often vascularized
- In widely invasive carcinoma, multiple satellite nodules display similar morphology
- Laboratory tests show absence of hypothyroidism
- It is proposed that dyshormonogenetic goiter be excluded in every case of follicular carcinoma diagnosed in a patient < 20 years (Int J Surg Pathol July 1999;7:125)
- Multifocal papillary thyroid carcinoma: cytologic, architectural (true papillae) and other (extrathyroidal extension, cervical or distant metastasis) features of cancer (Int J Surg Pathol July 1999;7:125)
- Poorly differentiated (insular) carcinoma: mitotic activity, necrosis, invasive growth
- Medullary thyroid carcinoma may resemble goiter with prominent myxomatosis and hyalinosis, but is calcitonin+
- Decreased T3, T4 and increased TSH
- Family history of hypothyroidism or goiter in 20% cases
- Highly atypical pleomorphic and hyperchromatic nuclei in internodular areas
- Increased T3, T4 and decreased TSH
- Markedly cellular nodules showing a variety of architectural patterns with minimal to absent pale colloid
Comment Here
Reference: Dyshormonogenetic goiter
- Capsular invasion of the nodules
- Highly atypical pleomorphic and hyperchromatic (bizarre) nuclei
- Large areas of necrosis
- Orphan Annie nuclei
- Scattered psammoma bodies
Comment Here
Reference: Dyshormonogenetic goiter

