
CNS & pituitary tumors
Gliomas, glioneuronal tumors and neuronal tumors
Adult type diffuse gliomas
Glioblastoma, IDH wild type
Deputy Editor-in-Chief: Chunyu Cai, M.D., Ph.D.


Last author update: 27 July 2022
Last staff update: 8 August 2024
Copyright: 2022-2025, PathologyOutlines.com, Inc.
PubMed Search: Glioblastoma, IDH wild type

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Gross description | Frozen section description | Intraoperative frozen / smear cytology images | Microscopic (histologic) description | Microscopic (histologic) images | Cytology description | Positive stains | Negative stains | Electron microscopy description | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Practice question #1 | Practice answer #1 | Practice question #2 | Practice answer #2Cite this page: Ramlal B, Morris M. Glioblastoma, IDH wild type. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/cnstumorgliomasglioblastomasidhwildtype.html. Accessed September 23rd, 2025.
Definition / general
- An aggressive, infiltrating, astrocytic glioma that lacks mutations in IDH1, IDH2 and histone H3 genes and is:
- Histologically defined by brisk mitotic activity and microvascular proliferation or necrosis
- Or molecularly defined by the presence of TERT promoter mutation, EGFR gene amplification or copy number changes in the form of combined gain of chromosome 7 and loss of chromosome 10 (Acta Neuropathol 2018;136:793, Acta Neuropathol 2018;136:805)
Essential features
- Characterized by diffusely infiltrative growth pattern with nuclear atypia and either:
- Mitotic activity, necrosis or microvascular proliferation or
- Molecularly defined by the present of TERT promoter mutation, EGFR amplification or combined gain of chromosome 7 and loss of chromosome 10
- Lacks mutations in IDH1 / IDH2 and histone H3 genes
- Various morphologic subtypes have been recognized (giant cell, small cell, epithelioid, sarcomatous / gliosarcoma) with similar prognosis
- Primitive neuronal component has increased rate of cerebrospinal fluid dissemination (Brain Pathol 2009;19:81)
Terminology
- Glioblastoma, IDH wild type
- Glioblastoma multiforme (not recommended)
- Diffuse astrocytoma with molecular features of glioblastoma (no longer recommended)
ICD coding
- ICD-O: 9440/3 - glioblastoma, NOS
- ICD-11: 2A00.00 & XH5571 - glioblastoma of brain & glioblastoma, IDH wild type
Epidemiology
- Most common and most malignant astrocytic glioma in adults (Pharmacol Res 2021;171:105780, Cancer Epidemiol Biomarkers Prev 2014;23:1985, StatPearls: Glioblastoma Multiforme [Accessed 5 July 2022])
- Accounts for 14.3% of all primary CNS tumors and 49.1% of all malignant CNS tumors in adults and up to 2.2% of all CNS tumors in children (Neuro Oncol 2021;23:iii1)
- More common in males than females (1.6:1)
- More common in older adults above the age of 55
- Highest incidence between the ages of 75 - 84 years
- Incidence rate by race: white to black = 1.98:1; white to Asian or Pacific Islander = 2.44:1
Sites
- Most commonly in supratentorial regions (frontal, temporal, parietal and occipital lobes), with highest incidence in the frontal lobes
- Most often centered in subcortical white matter
- Many cases show infiltration into cortex and across the corpus callosum with spread to contralateral hemisphere
- Rare cases reported in the cerebellum and spinal cord (World Neurosurg 2013;80:e237, J Neurosurg Spine 2010;13:67, Neurooncol Adv 2021;3:vdab154)
Pathophysiology
- Cell of origin undetermined
- Some studies suggest a variety of CNS cell types can undergo malignant transformation with features of glioblastoma (GBM) (oligodendrocyte precursor cells, neural precursor cells, astrocytes and neurons)
- Sequencing of human glioblastomas suggests that a neural precursor cell in the subventricular zone may be the cell of origin (Nature 2018;560:243)
Etiology
- Etiology of most GBMs remains unknown
- Rare cases associated with genetic tumor syndromes: Lynch syndrome, Li-Fraumeni syndrome, tuberous sclerosis and neurofibromatosis type 1 (Neuro Oncol 2014;16:896)
- Only validated risk factor is ionizing radiation to the head and neck (Neurooncol Adv 2021;3:vdab109, Neuro Oncol 2002;4:278, UpToDate: Risk Factors for Brain Tumors [Accessed 5 July 2022])
- Some radiation associated gliomas with histologic features of glioblastoma may be molecularly more consistent with diffuse pediatric type high grade glioma, H3 wild type and IDH wild type (Acta Neuropathol 2019;137:139)
- Conflicting evidence over a possible association with traumatic brain injury (TBI) (Surg Neurol Int 2016;7:78, J Int Med Res 2018;46:2170, Cureus 2020;12:e8019)
Clinical features
- Symptoms are dependent on tumor location
- May present with signs of increased intracranial pressure (i.e., headaches, nausea, emesis), seizures or focal neurologic deficits (visual field defects, hemiparesis, aphasias, etc.) (BMJ 2021;374:n1560)
Diagnosis
- Computed tomography (CT) and magnetic resonance imaging (MRI) for radiologic assessment
- Biopsy or surgical resection required for definitive diagnosis
- May be able to detect glioblastoma by circulating tumor DNA (ctDNA) in the blood and cerebrospinal fluid (CSF) of some patients, though this is under investigation (J Neurosurg 2018;129:334, Cancers (Basel) 2019;11:950, Mol Oncol 2022;16:2098, Cancers (Basel) 2020;12:1219)
Radiology description
- MRI: T2 / fluid attenuated inversion recovery (FLAIR) bright infiltrative lesion(s) with postcontrast T1 showing irregular peripheral rim enhancement with central necrosis
- May cross the corpus callosum
- May be multifocal
- Lack of contrast enhancement may be observed in molecularly defined glioblastoma
- Certain subtypes (i.e., gliosarcoma, epithelioid, giant cell) may appear well circumscribed (Neurosurg Rev 2021;44:3335, Eur Radiol 2019;29:429, Surg Pathol Clin 2020;13:249)
Radiology images
Contributed by Bharat Ramlal, M.D.
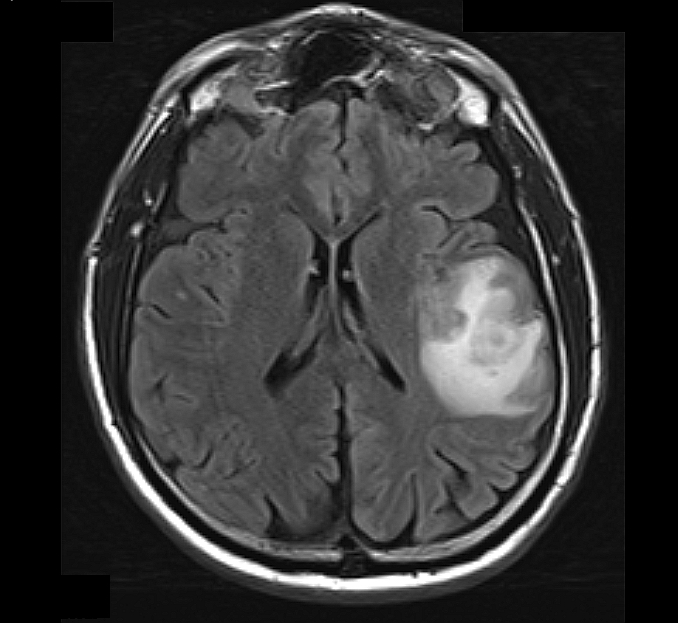
T2 FLAIR MRI
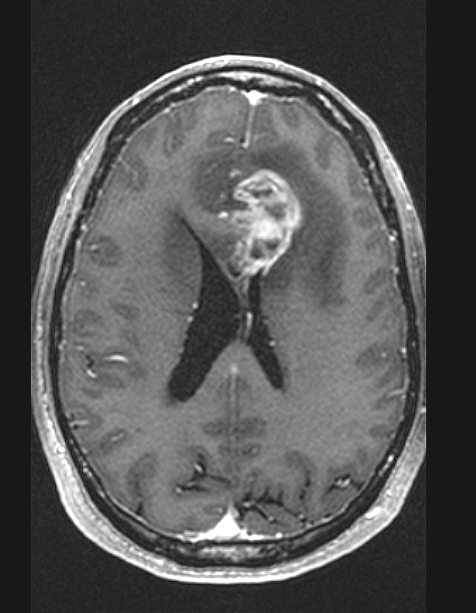

T1 postcontrast MRI
Prognostic factors
- Poor prognosis, with a median survival of 8 months and 5 year survival rate of only 6.8% (Neuro Oncol 2021;23:iii1)
- Most patients die within 15 - 18 months after therapy with chemoradiation
- Longer survival is observed in patients who are diagnosed at a younger age (< 50 years), have high performance status and gross total resection (often difficult) (J Clin Oncol 2009;27:5743)
- MGMT promoter methylation is a positive predictor of response to alkylating and methylating chemotherapy and may be an independent prognostic factor for longer overall survival (J Cell Physiol 2018;233:378, J Clin Oncol 2009;27:5743)
- Brisk cytotoxic T cell infiltrates may be associated with longer survival (J Clin Neurosci 2010;17:1381)
- FGFR3::TACC3 fusion GBMs may be associated with better outcomes and may be a target for therapy (Neuro Oncol 2020;22:1614, Clin Cancer Res 2015;21:3307)
- Shorter survival times than patients with IDH mutant grade 4 astrocytomas with similar histological findings (Clin Cancer Res 2013;19:5146, Acta Neuropathol 2018;136:153)
Case reports
- 46 year old man with glioblastoma and subsequent scalp and pulmonary metastases (J Int Med Res 2020;48:300060520930459)
- 47 year old woman with primary intraventricular epithelioid glioblastoma (World Neurosurg 2018;112:257)
- 47 year old man with traumatic brain injury secondary to a fall and subsequent development of GBM (Cureus 2020;12:e8019)
- 76 year old woman with primary glioblastoma of the cauda equina (Neurooncol Adv 2021;3:vdab154)
Treatment
- Surgical resection where possible in younger patients (≤ 70 years old) and patients with good performance status, followed by radiotherapy with concurrent and adjuvant temozolomide (TMZ)
- Unmethylated tumors, standard brain radiotherapy alone may be attempted (NCNN: NCNN Guidelines - Central Nervous System Cancers [Accessed 5 July 2022])
- Tumor treating fields (TTFields / Optune) under investigation - alternating electric field therapy using low intensity energy to stop glioma proliferation; relatively recent treatment option with rare reports showing favorable outcomes (Front Oncol 2020;10:477)
Gross description
- Ill defined whitish gray mass with areas of hemorrhage and necrosis
- Can expand gyri and cross the corpus callosum
Frozen section description
- Hypercellular infiltrative lesion with variable morphology
- Infiltration often difficult to assess on frozen sections but entrapped neurons may be useful
- Nuclear hyperchromasia and nuclear elongation, possible giant cells
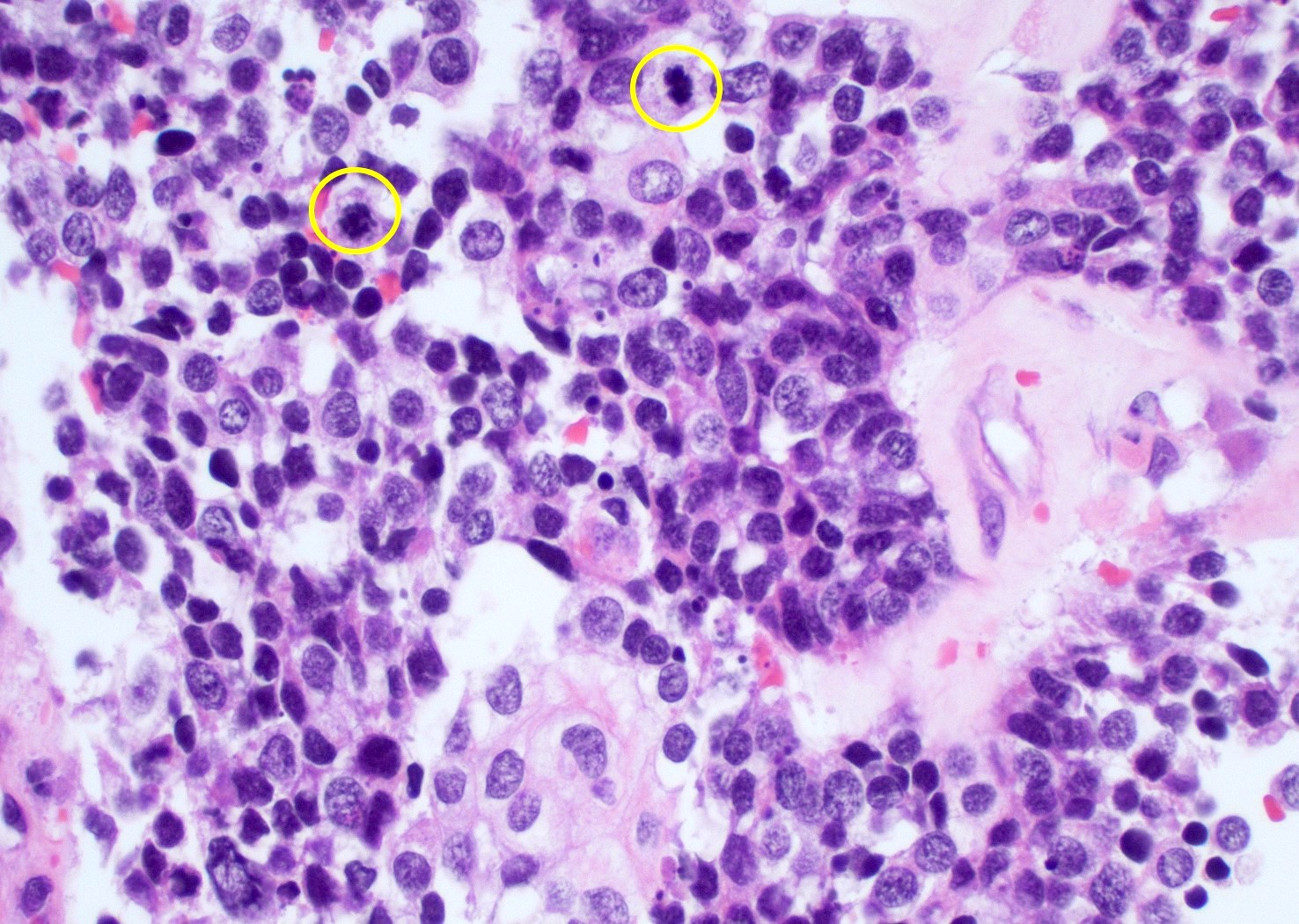
- Mitotic activity
- Necrosis; geographic or pseudopalisading
- Microvascular proliferation
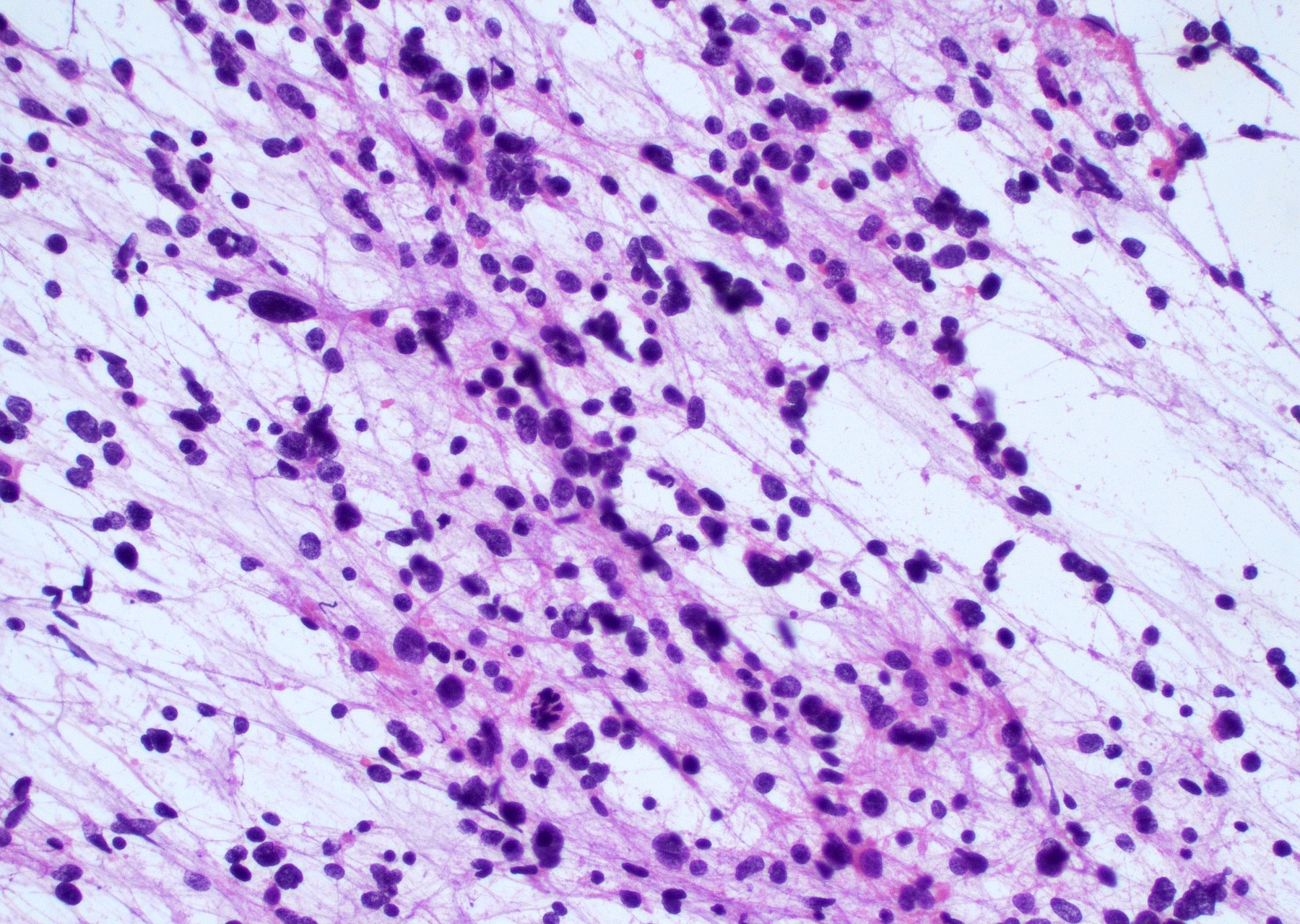
Intraoperative frozen / smear cytology images
Contributed by Bharat Ramlal, M.D.
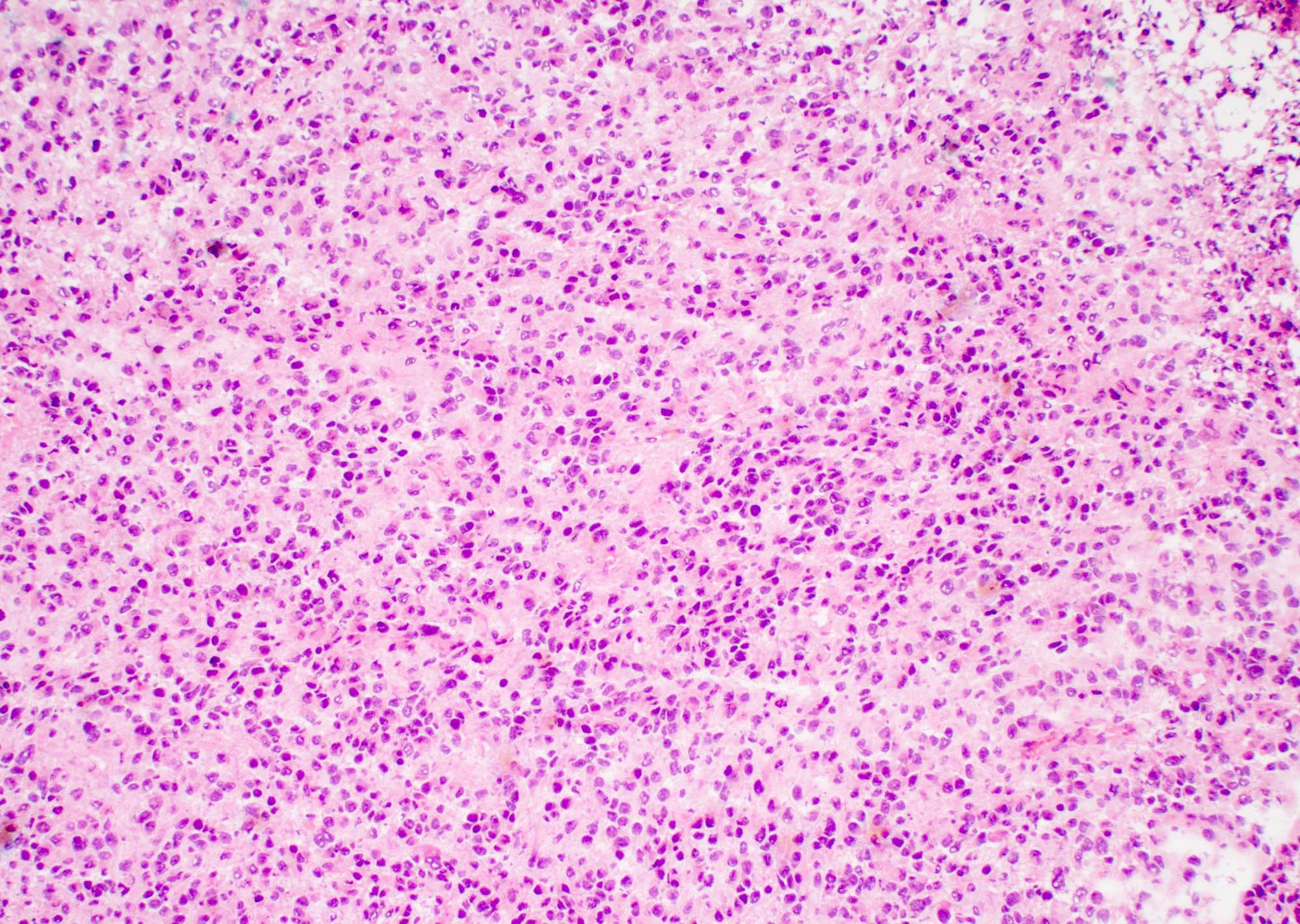
Markedly hypercellular neoplasm
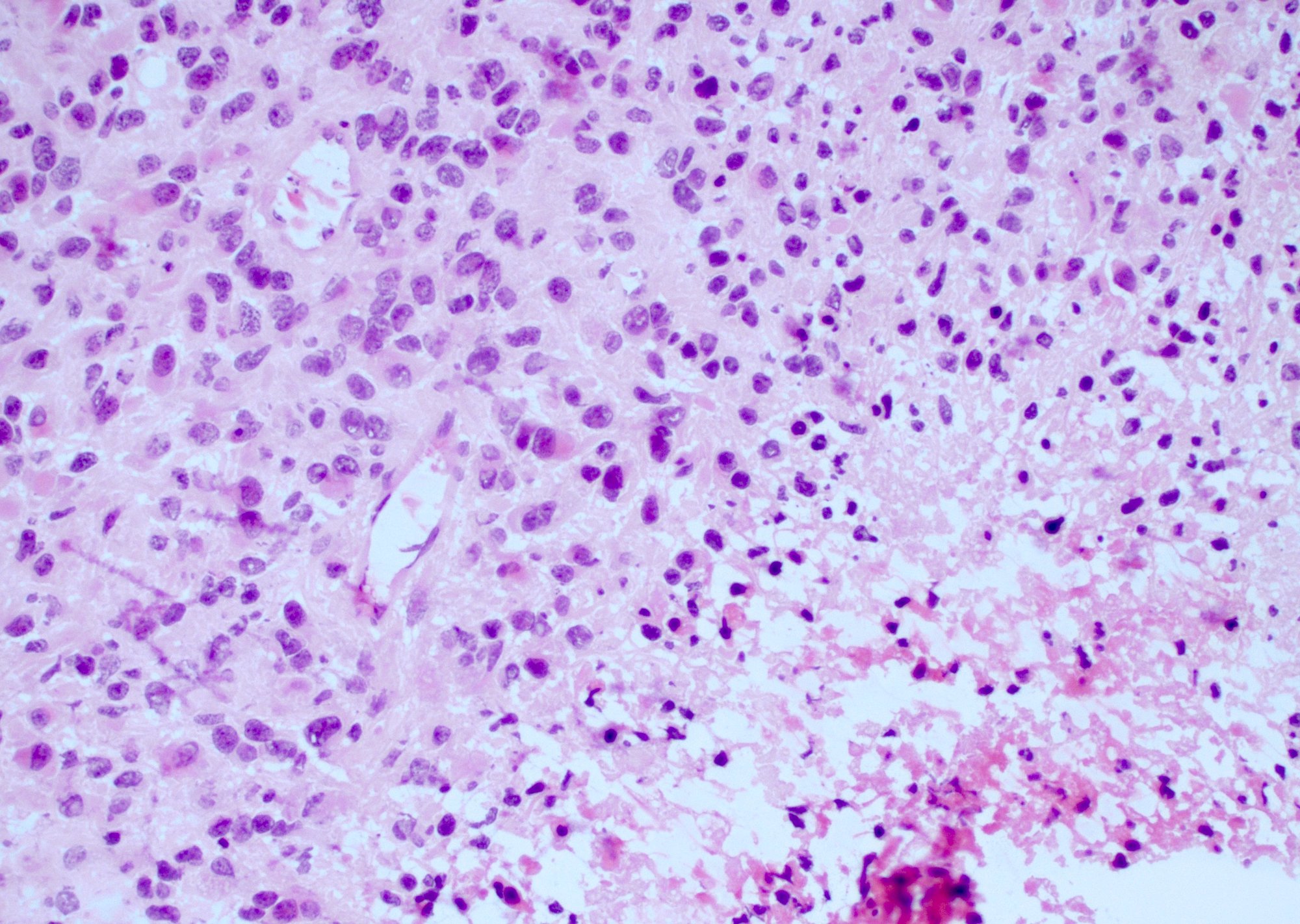
Nuclear atypia and palisading tumor cells
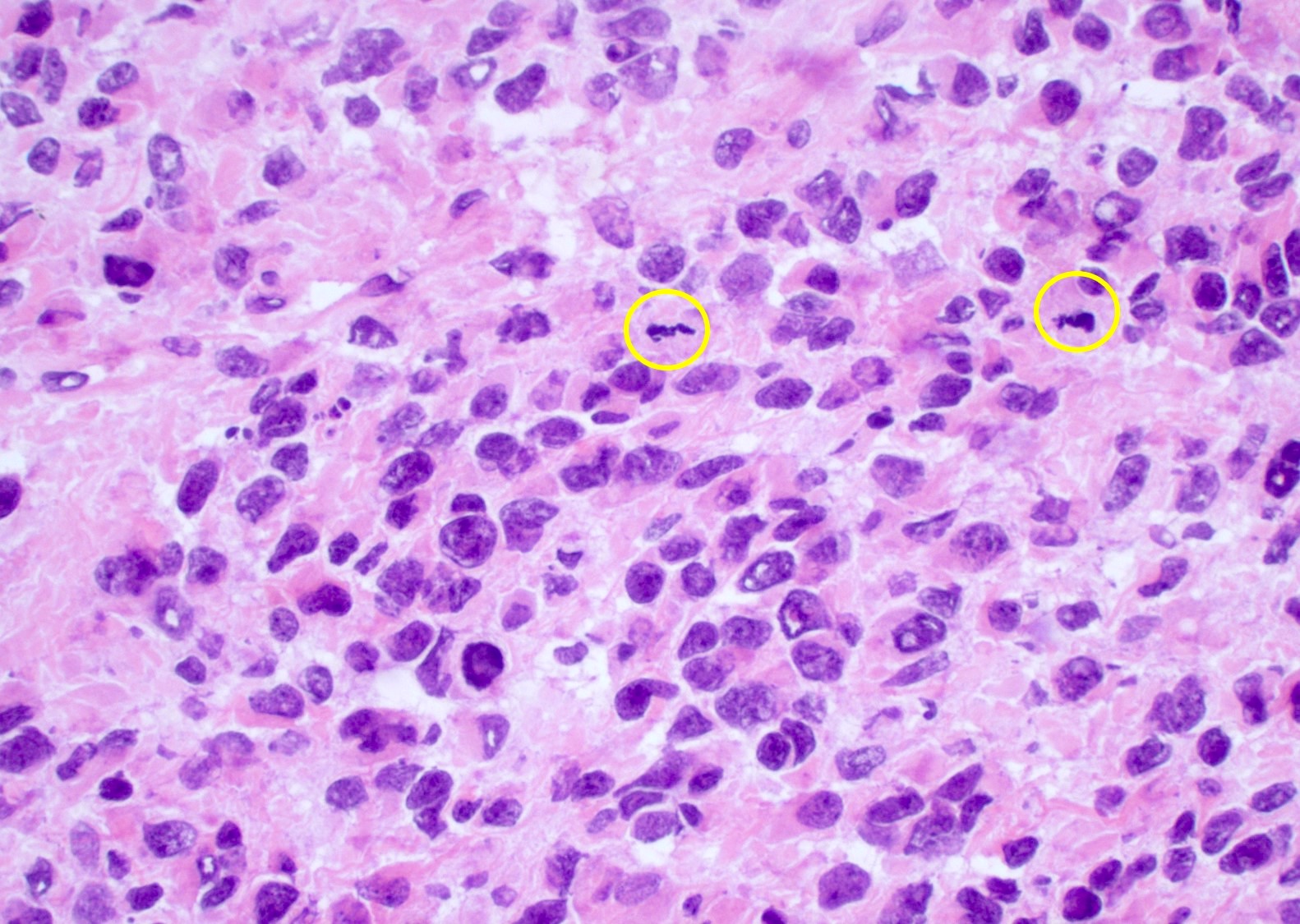
Pleomorphic, astrocytic tumor cells
Intraoperative smear preparation
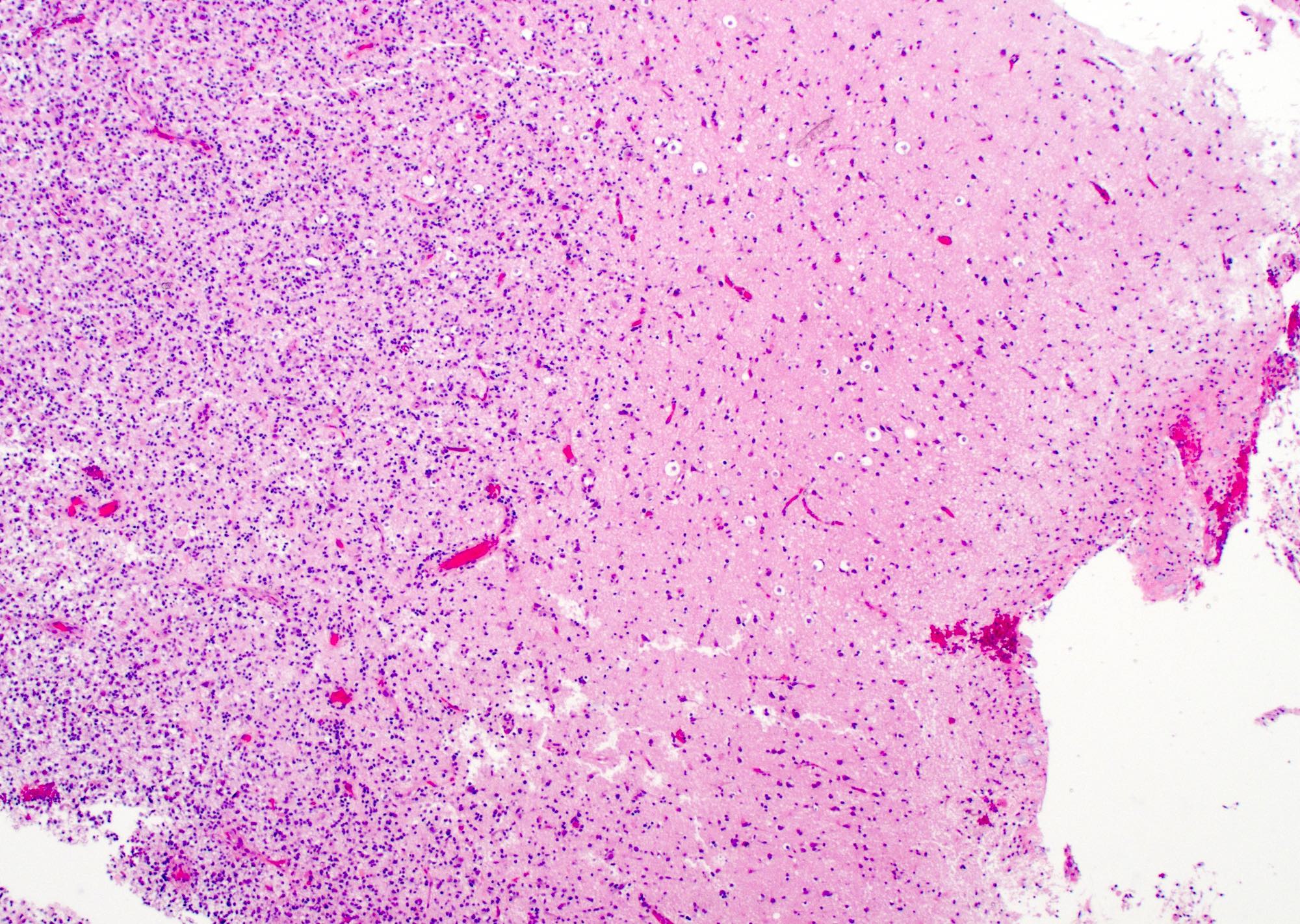
Microscopic (histologic) description
- Infiltrating, hypercellular astrocytic neoplasm often with hyperchromatic, elongated nuclei and irregular nuclear membranes
- Typically mitotically active, though not required if molecular criteria are met
- Microvascular proliferation or necrosis is required for a histologic diagnosis of GBM
- Microvascular proliferation: multilayered, small caliber vessels with glomeruloid appearance (J Neuropathol Exp Neurol 1992;51:488)
- Necrosis: can be geographic or pseudopalisading with neoplastic cells surrounding central necrosis
- Greater association of thrombosis and necrosis in IDH wild type GBM than in IDH mutant grade 4 astrocytomas (Acta Neuropathol 2016;132:917)
- Variable cell morphology: undifferentiated / primitive neuronal cells, astrocytic, gemistocytic, oligodendroglial-like, small cell, lipidized, granular, epithelioid, giant cells, mesenchymal metaplasia and epithelial metaplasia
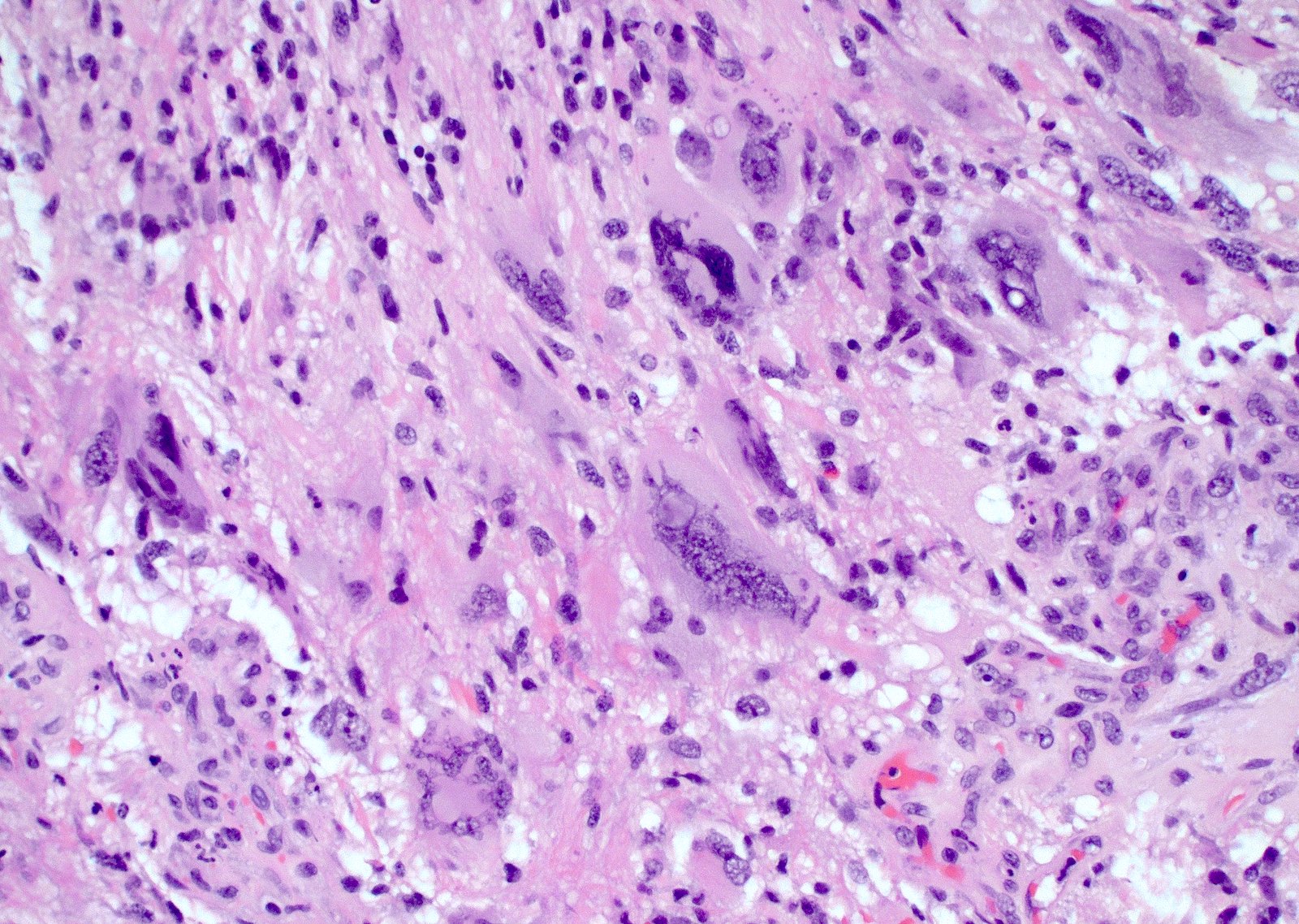
- Primitive neuronal cells (embryonal): markedly increased cellularity composed of cells with high N/C ratio, brisk mitotic activity with apoptotic bodies, nuclear molding, sometimes with neuroblastic rosettes
- Typically has conventional infiltrating astrocytic component, which is morphologically distinct
- Loss of glial markers, expression of neuronal markers (synaptophysin)
- Higher risk of CSF dissemination but similar survivals as classic GBM
- Associated with MYC amplifications
- Astrocytic: fibrillary, elongated processes
- Gemistocytic: abundant eosinophilic cytoplasm with eccentric nuclei
- Oligodendroglial-like: cells with small, round nuclei with perinuclear clearing in a vascular background
- Can be associated with FGFR3::TACC3 fusion positive GBM (Brain Pathol 2018;28:674)
- Small cell change: monomorphic cells with small, round or angulated, hyperchromatic nuclei and brisk mitotic activity
- Associated with EGFR amplification (Clin Neuropathol 2005;24:163, J Neuropathol Exp Neurol 2001;60:1099)
- Lipidized / xanthomatous cells: cells with abundant foamy cytoplasm
- Be sure to exclude pleomorphic xanthoastrocytoma
- Granular cells: large cells with small nuclei and abundant granular cytoplasm
- PAS positive
- May be CD68 positive but negative for CD163
- Epithelioid: large eosinophilic cells with prominent nucleoli
- May resemble rhabdoid cells with more eccentric nuclei
- May be immunoreactive to cytokeratins but negative for CAM5.2
- May be more sharply demarcated with less infiltration
- Often associated with BRAF V600E mutations, usually in younger individuals (Discov Med 2018;26:51, Int J Clin Exp Pathol 2020;13:1529)
- Giant cell: well circumscribed tumors composed of markedly pleomorphic and bizarre cells, including multinucleated tumor cells
- Often have a rich reticulin network
- Associated with mutations in TP53, RB1, NF1 and POLE (Neurooncol Adv 2020;2:vdz059, Acta Neuropathol Commun 2021;9:200, Neuro Oncol 2015;17:1356)
- POLE mutated tumors associated with ultramutated phenotype and may have a better prognosis
- Mesenchymal / sarcomatous: may be well circumscribed; corresponds to cellular differentiation along various lineage; sarcomatous (spindled and fibroblastic), osseous, chondroid or myogenic differentiation (see Gliosarcoma)
- Sarcomatous component usually comprised of GFAP negative spindled cells with reticulin deposition rich
- Associated with mutations in TP53, PTEN, TERT and CDKN2A deletion and MDM2 and CDK4 coamplification (J Neuropathol Exp Neurol 1995;54:651, Am J Pathol 2000;156:425, Acta Neuropathol 2001;101:321)
- Epithelial metaplasia: rare but may include squamous or adenomatous differentiation
- Keratin pearls, epithelial whorls: CK5/6 positive
- Glandular structures
- Primitive neuronal cells (embryonal): markedly increased cellularity composed of cells with high N/C ratio, brisk mitotic activity with apoptotic bodies, nuclear molding, sometimes with neuroblastic rosettes
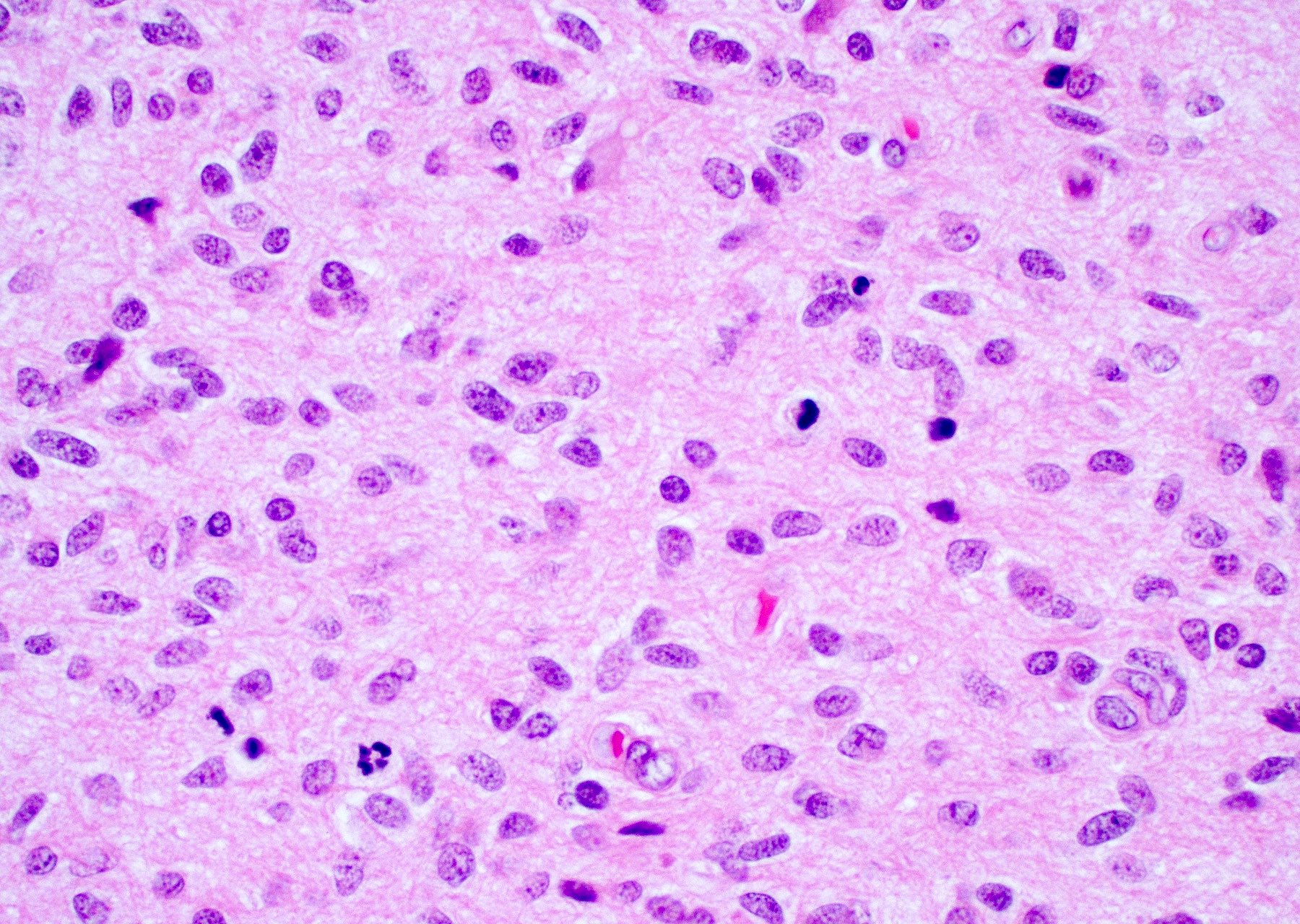
Microscopic (histologic) images
Contributed by Bharat Ramlal, M.D. and Meaghan Morris, M.D., Ph.D.
Hypercellular neoplasm
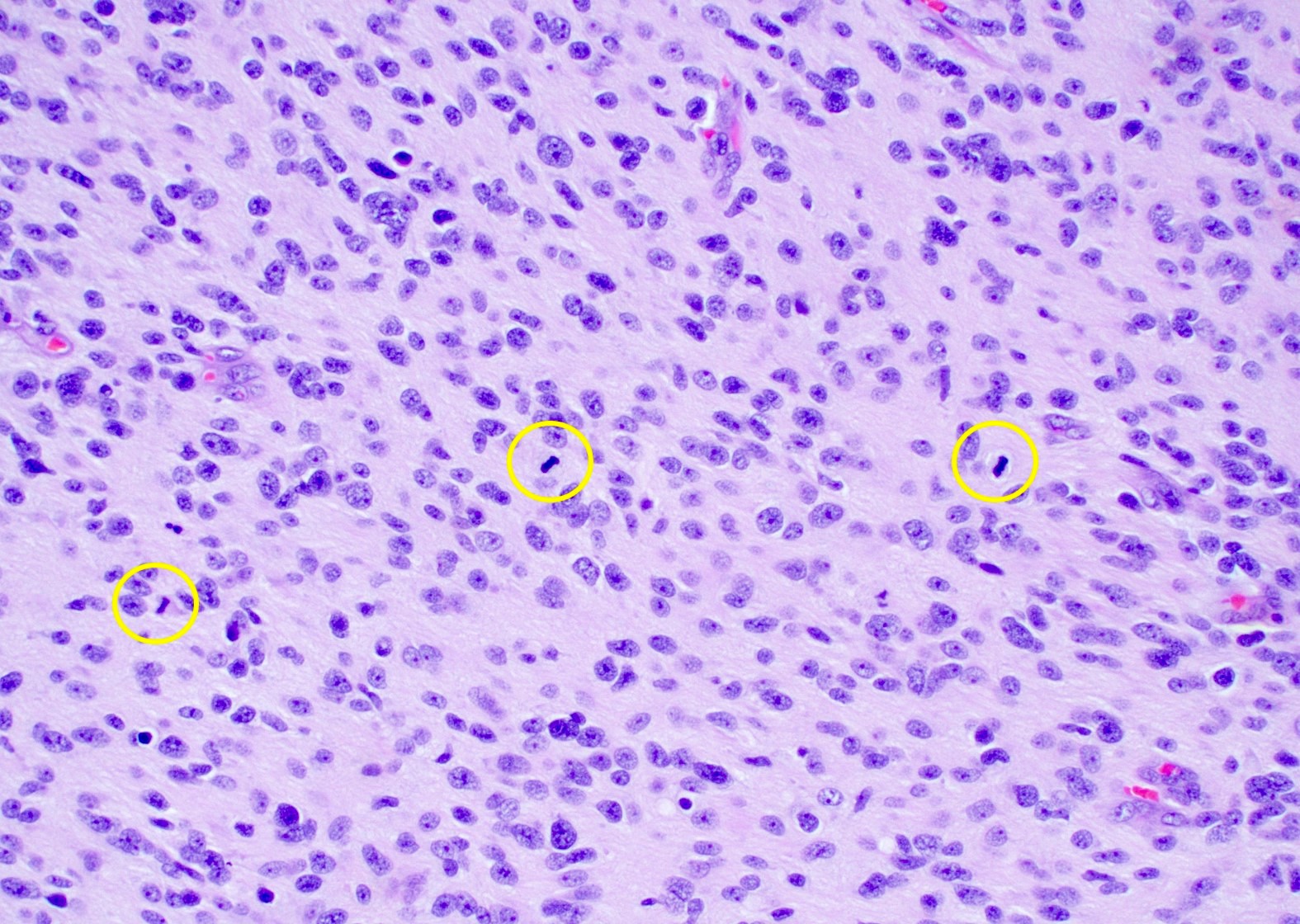
Nuclear atypia and brisk mitotic activity
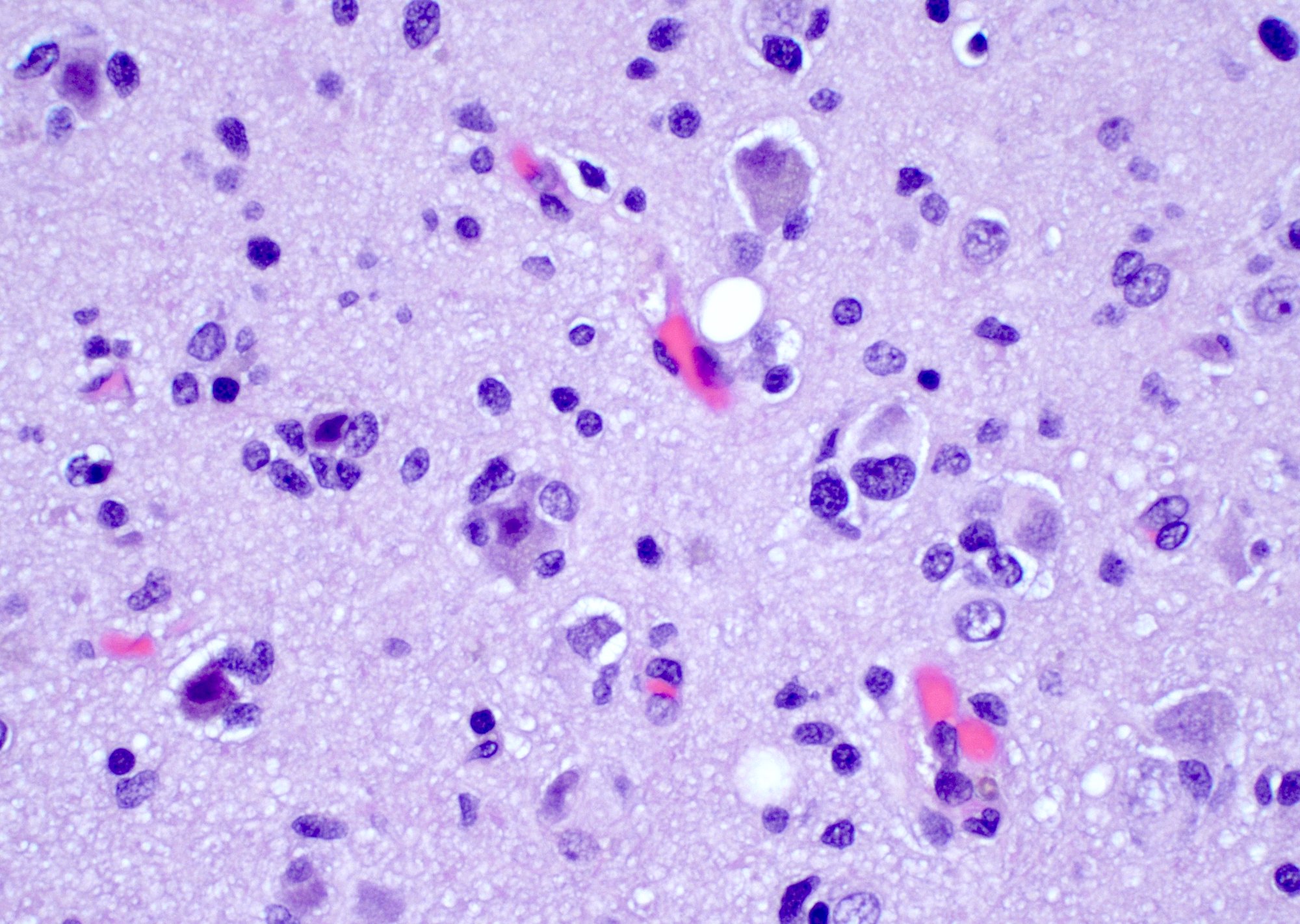
Atypical astrocytic tumor cells
Glomeruloid microvascular proliferation

Pseudopalisading necrosis
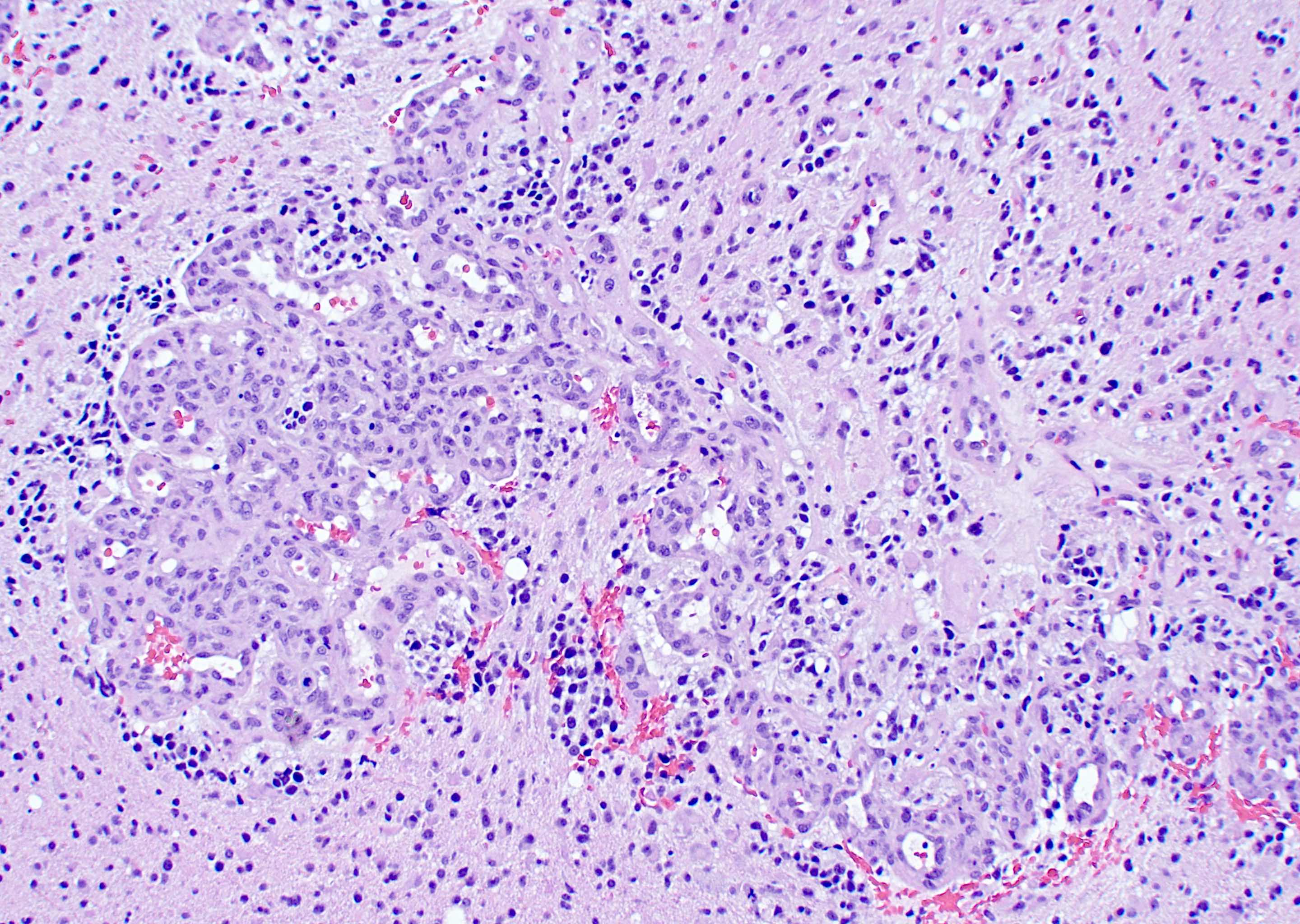
Marked nuclear pleomorphism
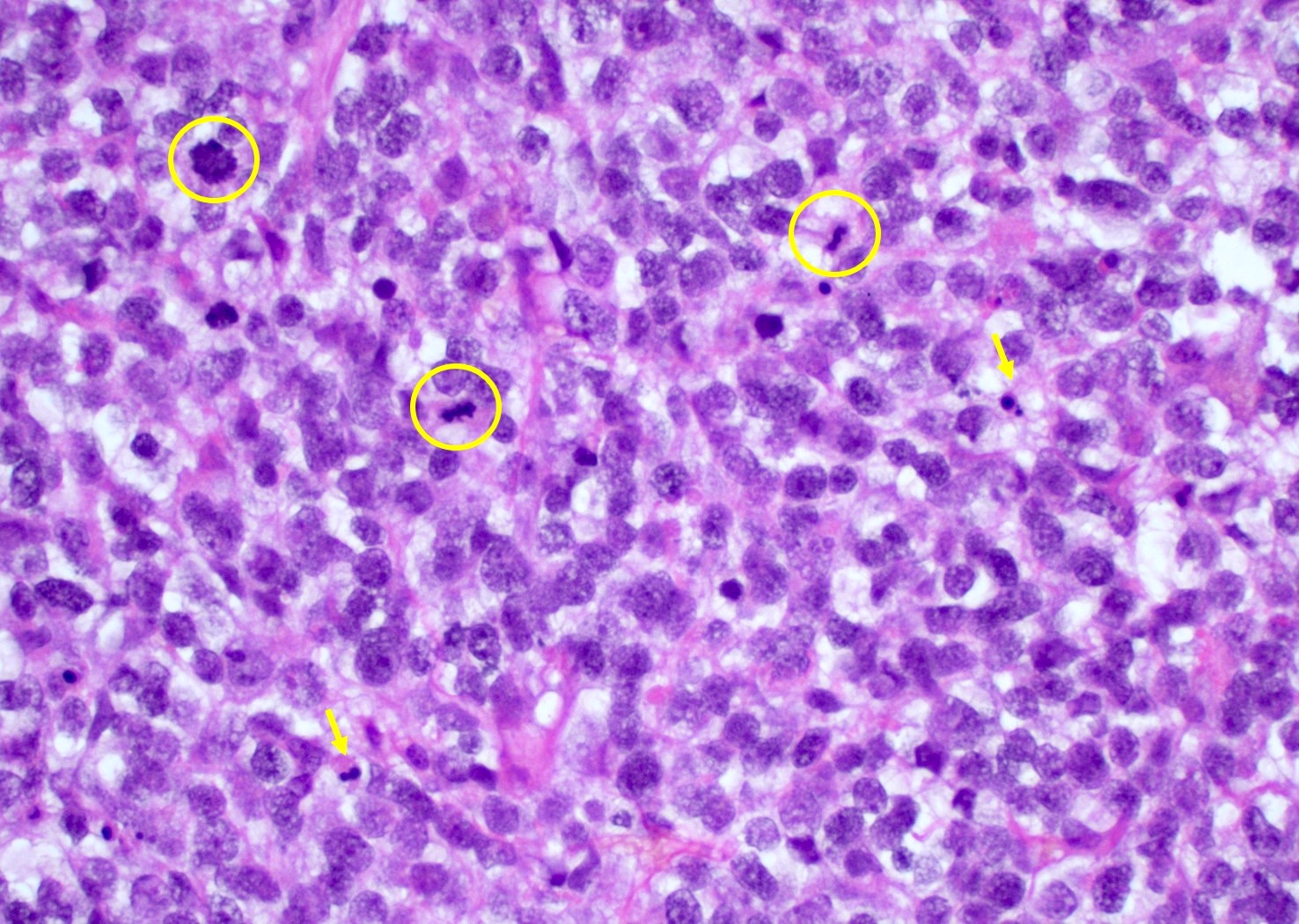
Primitive neuronal component
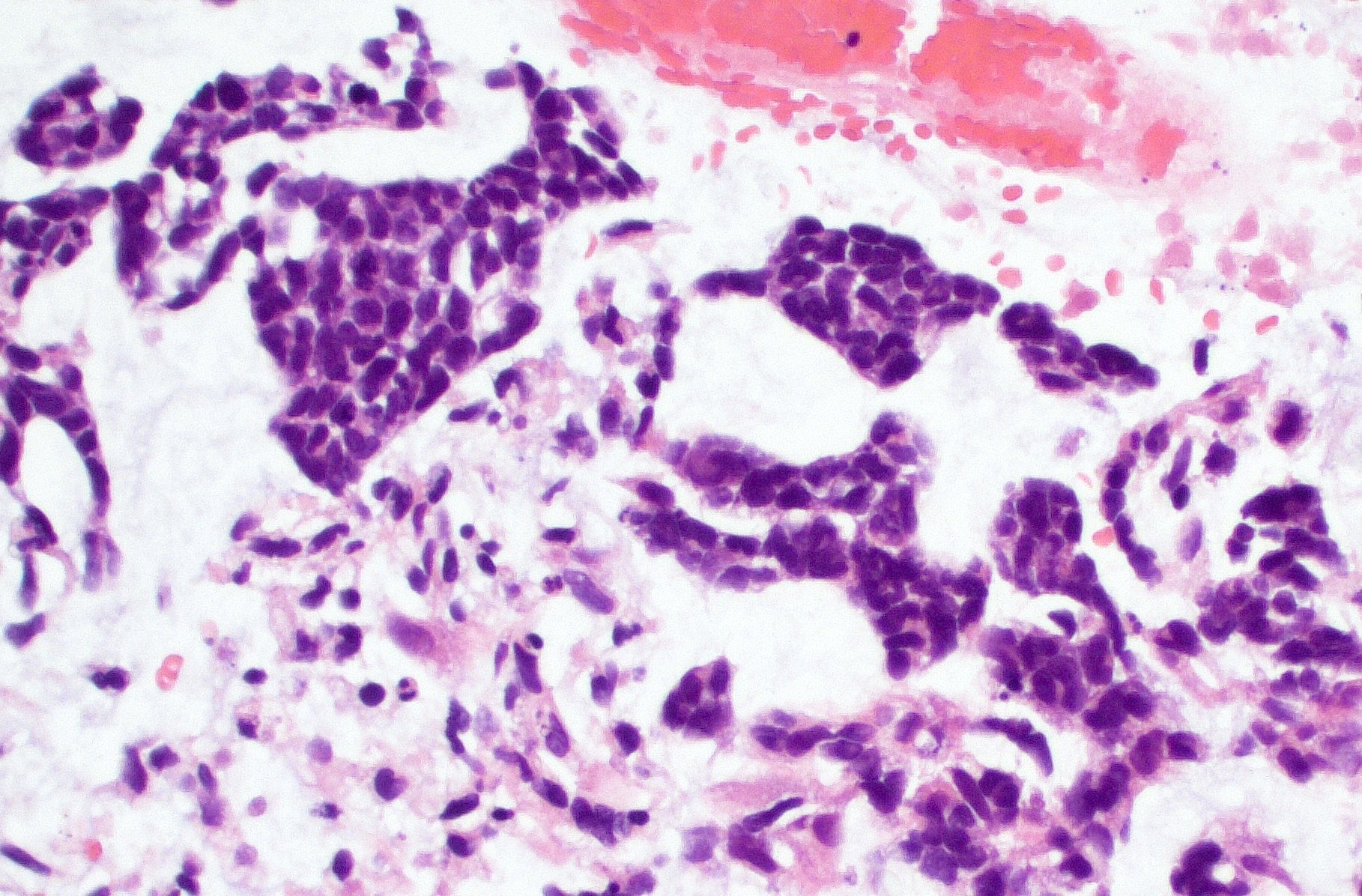
Adenoid morphology

Epithelioid glioblastoma
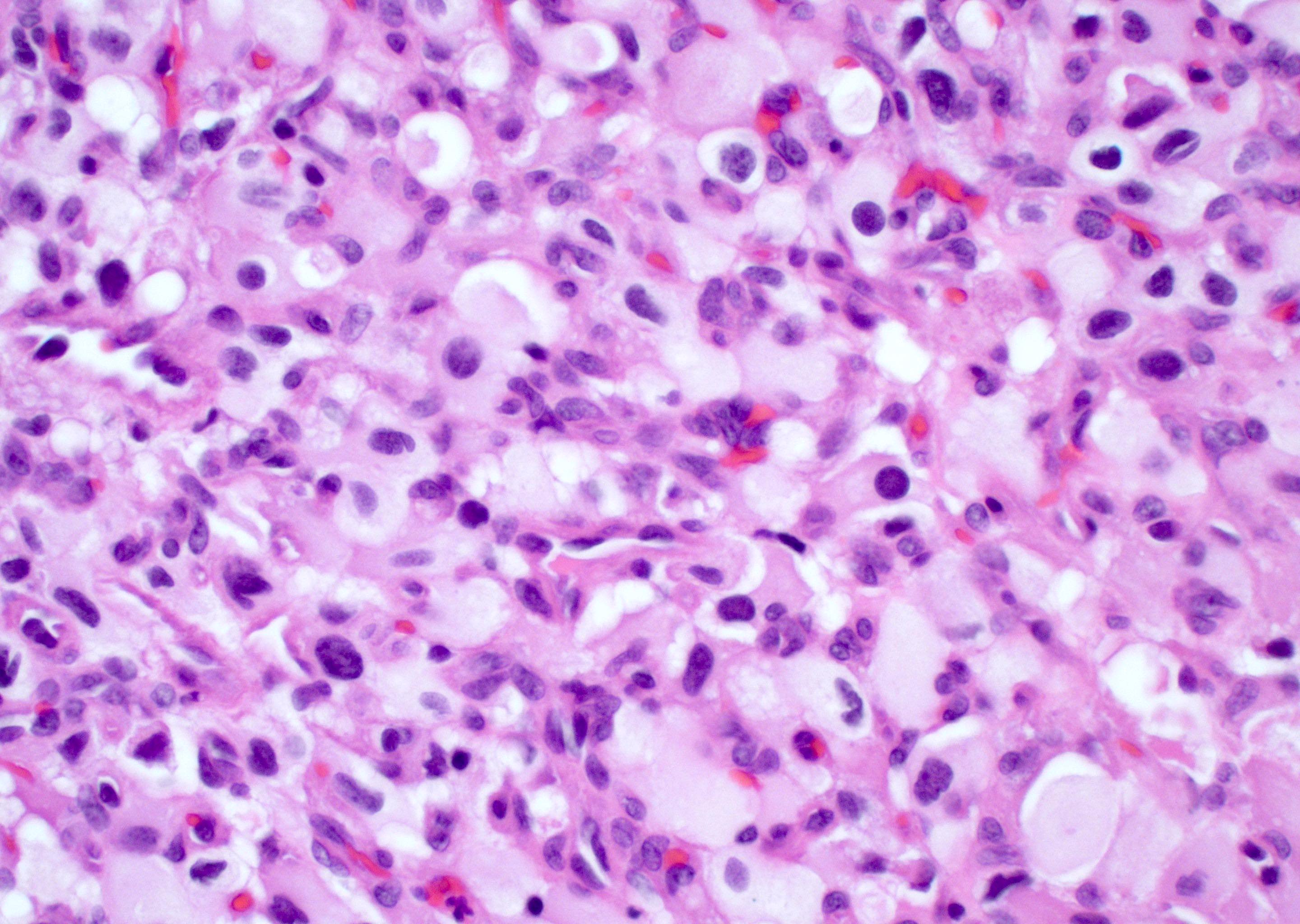

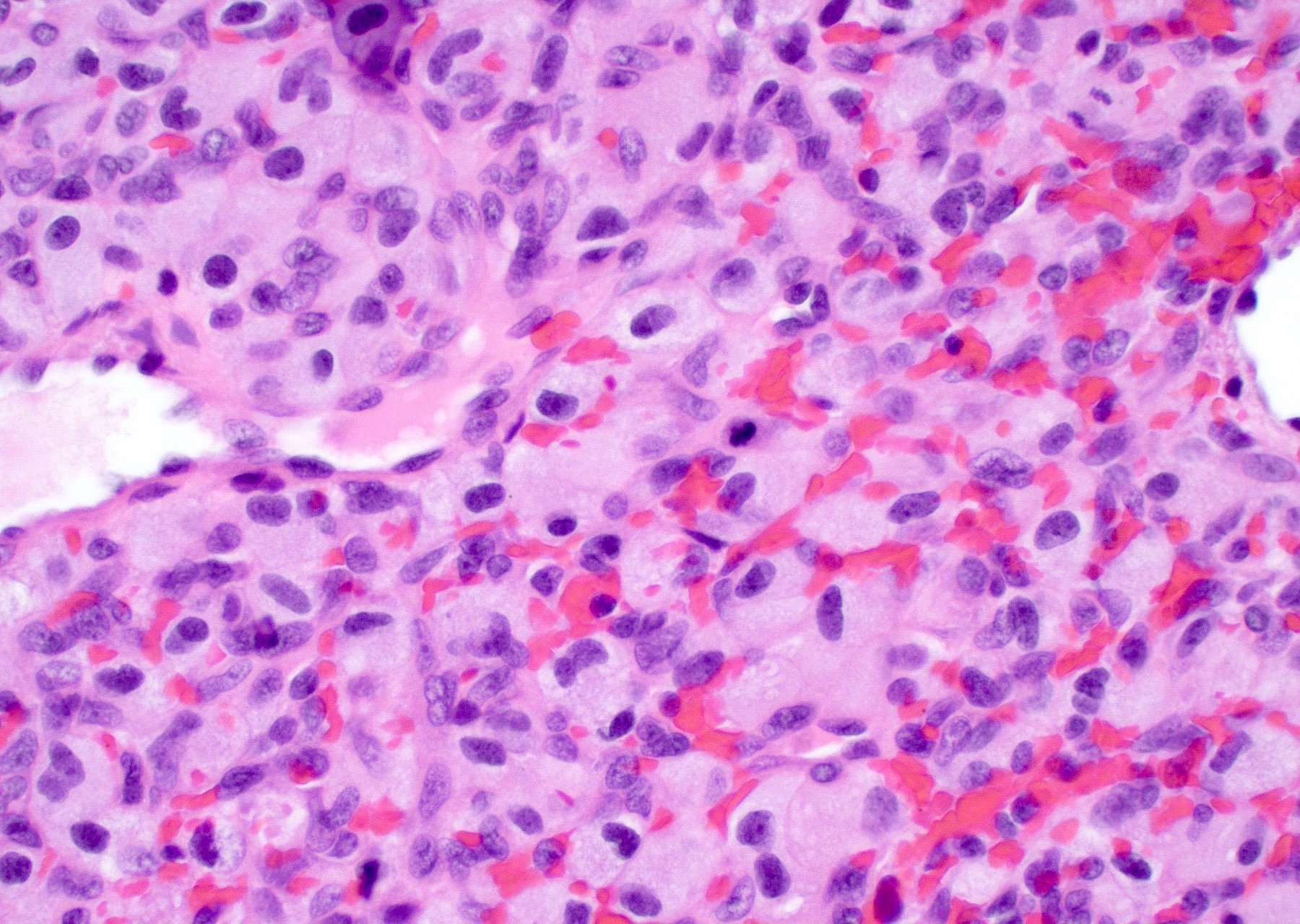
Granular cell morphology
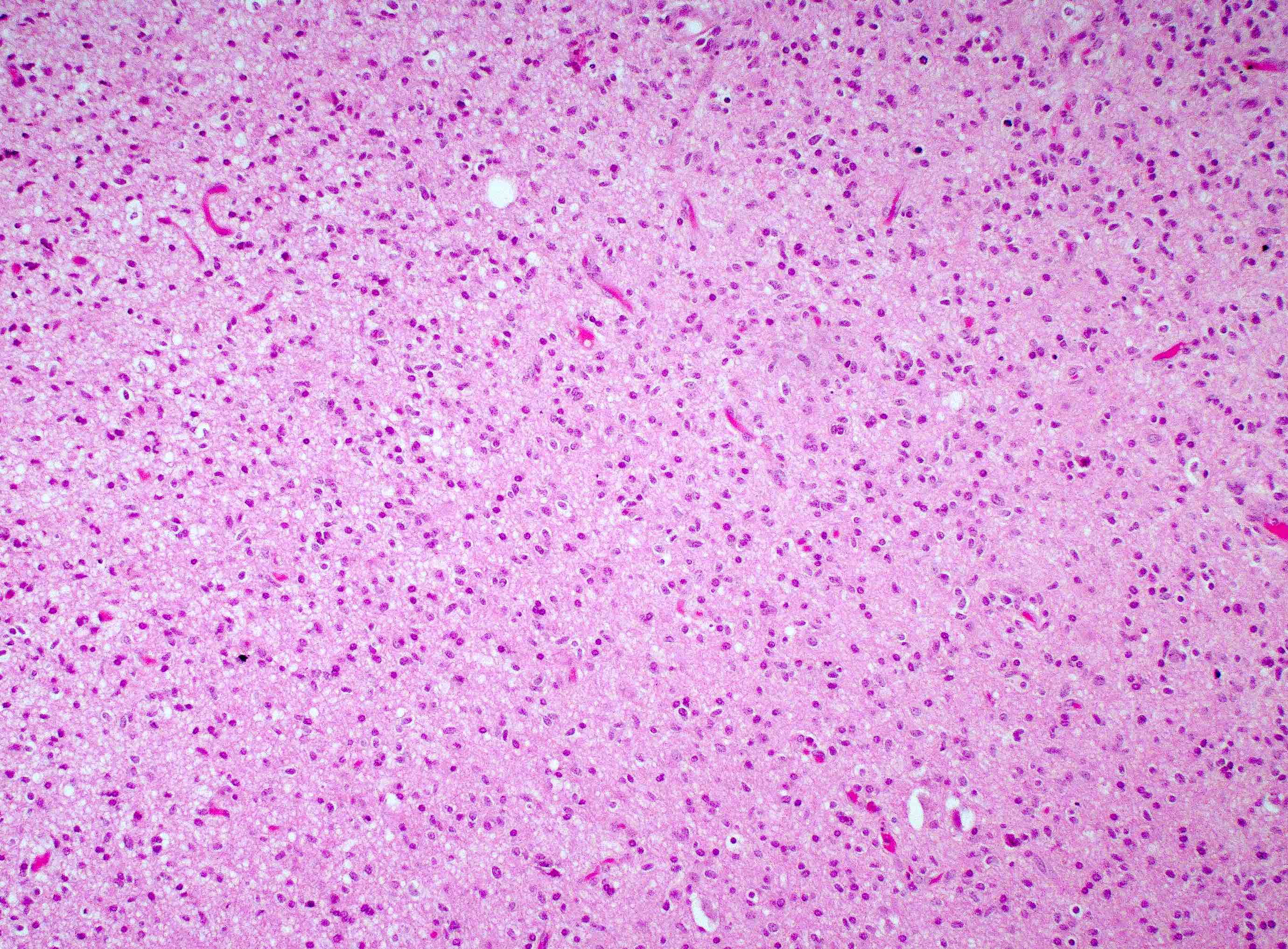
Small cell glioblastoma

IDH1 R132H
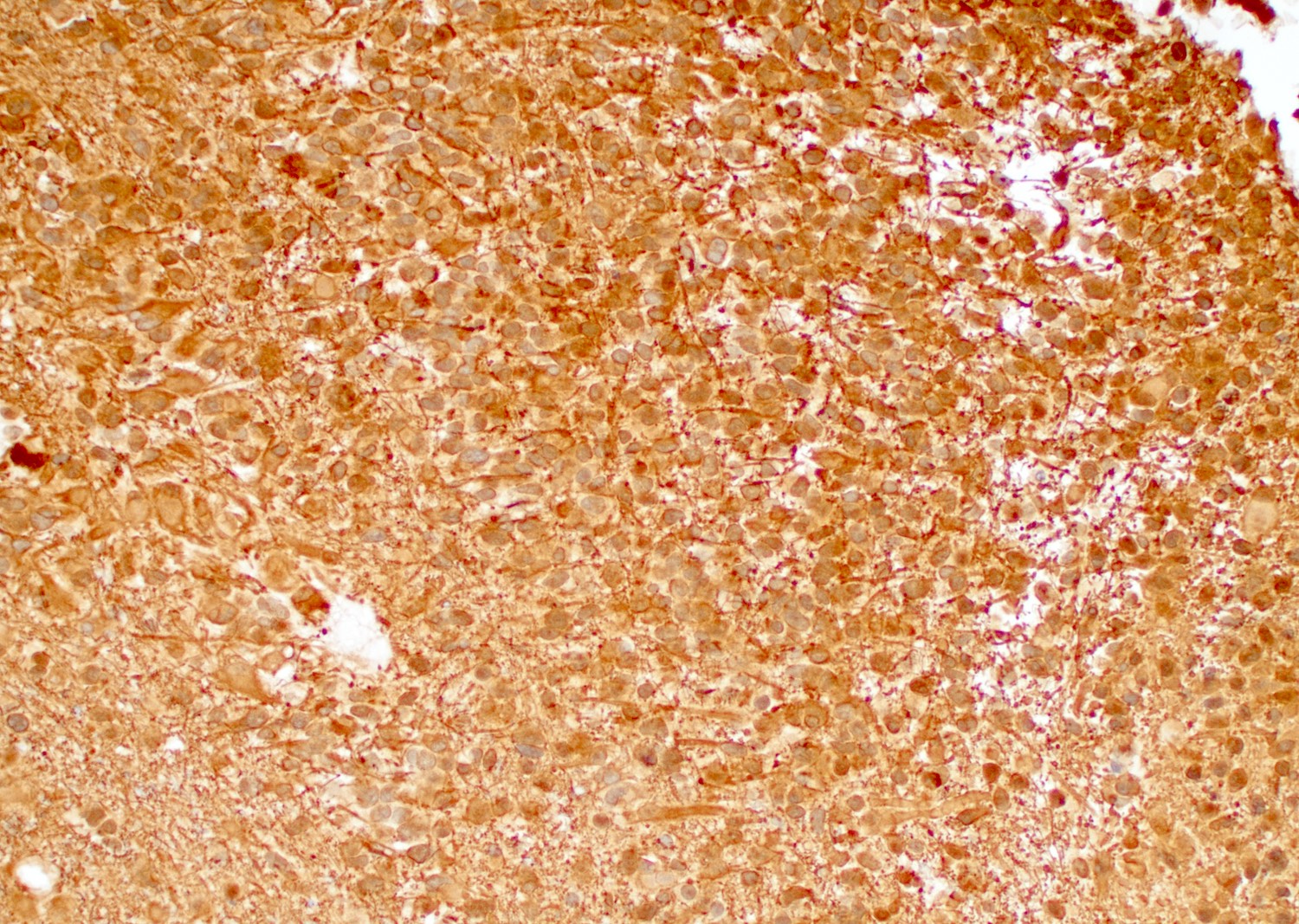
GFAP
Olig2

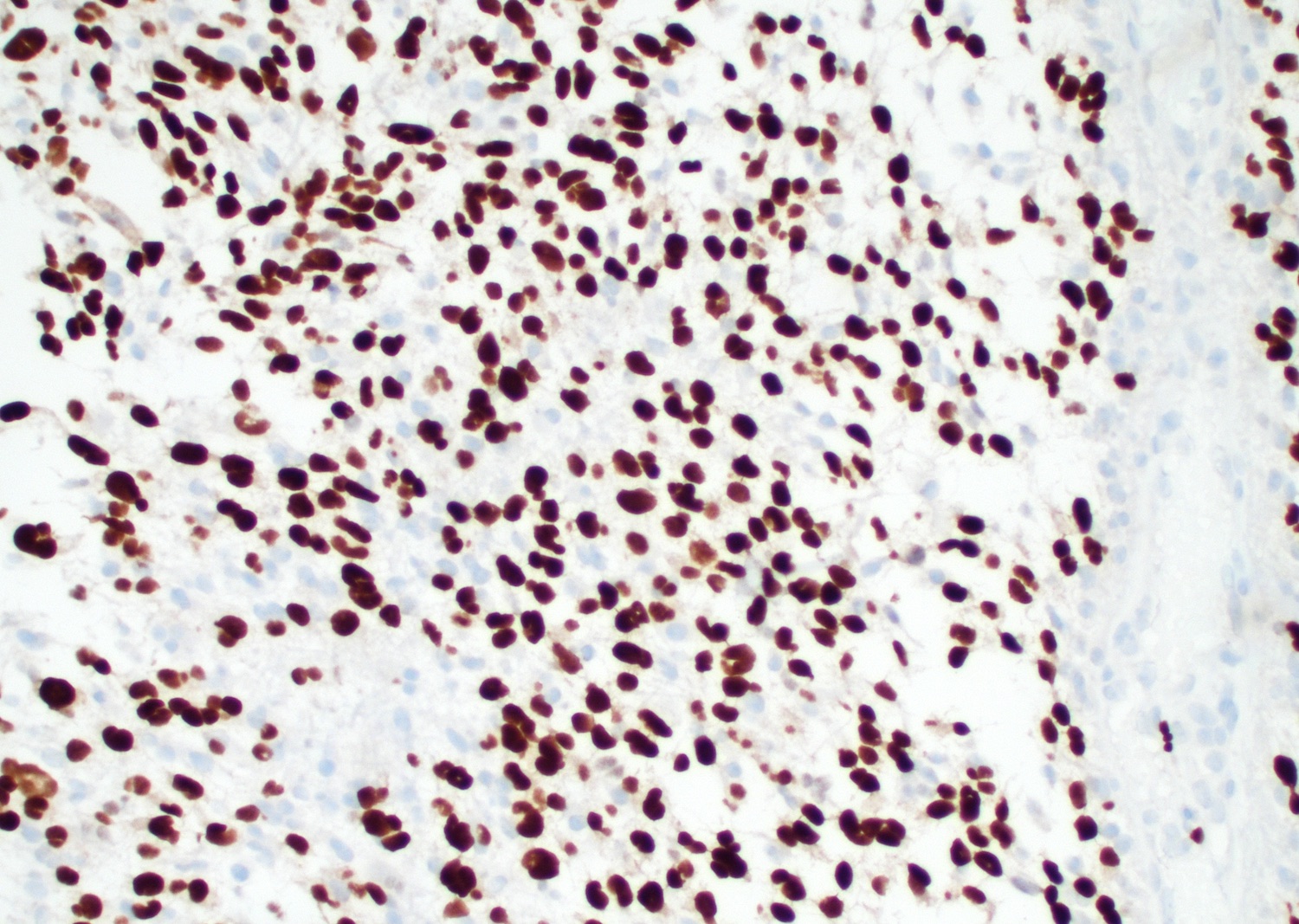
Ki67

Synaptophysin
Cytology description
- Intraoperative smears may show marked cellularity, with moderate to markedly pleomorphic astrocytic / gemistocytic cells with fine fibrillar glial processes (Diagn Cytopathol 1986;2:312)
- May show necrosis
- Mitotic figures can be observed
Positive stains
- GFAP, Olig2, S100
- GFAP may be lost in GBMs with sarcomatoid or primitive neuronal components
- Cytokeratin AE1 / AE3 may cross react with glial filaments (Pathol Oncol Res 2015;21:817)
- ATRX and H3K27 trimethylation usually retained
- EGFR (Indian J Med Res 2017;146:738)
- Nuclear p53 in up to 30% of GBM; more common in giant cell GBM
- BRAF V600E in epithelioid GBMs (Am J Surg Pathol 2015;39:528)
- Synaptophysin, chromogranin, INSM1 in primitive neuronal component; usually negative in glioblastoma lacking a primitive neuronal component
- Ki67 usually high; can be low in molecularly defined GBMs
Negative stains
- IDH1 (R132H mutant protein) (Acta Neuropathol 2016;131:803)
- H3 (K27M mutant protein) (Acta Neuropathol 2016;131:803, Ann Pathol 2021;41:137)
- H3 (G34R mutant protein) (Acta Neuropathol 2016;131:803, Brain Tumor Pathol 2021;38:4)
Electron microscopy description
- Bundles of cytoplasmic filaments 80 - 90 angstroms in diameter (Am J Pathol 1973;73:589)
- Pleomorphic nuclei and prominent nucleoli with nuclear infoldings and cytoplasmic invaginations (intranuclear pseudoinclusions)
Molecular / cytogenetics description
- Lacks mutations in IDH1 / IDH2 and histone H3 genes
- Lack of IDH1 immunohistochemistry sufficient in patients ≥ 55 years of age meeting histologic criteria for glioblastoma with nonmidline tumors (Acta Neuropathol 2016;131:803, Oncotarget 2016;7:31393)
- Molecularly defined GBM: even in low grade appearing tumors and tumors lacking necrosis or microvascular proliferation (Acta Neuropathol 2018;136:805, Acta Neuropathol 2018;136:793):
- TERT promoter mutation
- EGFR gene amplification or combined whole chromosome 7 gain and whole chromosome 10 loss (+7 / -10)
- Alterations of RTK genes, PI3K pathway genes and PTEN in up to 90% of GBM (Cell 2013;155:462, Nature 2008;455:1061)
- CDKN2A / CDKN2B deletion can be present
- BRAF V600E in up to 50% of GBMs with epithelioid features (Am J Surg Pathol 2013;37:685)
- MYC or MYCN amplification associated with primitive neuronal component
- Radiation induced gliomas: PDGFRA, TP53, CDKN2A deletion (Neurooncol Adv 2021;3:vdab109, Nat Commun 2021;12:5530, Acta Neuropathol 2019;137:139)
- Some radiation associated gliomas with histologic features of glioblastoma may be molecularly more consistent with diffuse pediatric type high grade glioma, H3 wild type and IDH wild type (Acta Neuropathol 2019;137:139)
- If present, gene fusions most commonly involve the receptor tyrosine kinase (RTK) family (Cell 2013;155:462, Cancers (Basel) 2020;12:1219, Front Oncol 2020;10:593578):
- EGFR
- FGFR3
- MET
- NTRK1 / NTRK2 / NTRK3
- PDGFRA
Sample pathology report
- Brain, right frontal lobe, biopsy:
- Glioblastoma, IDH wild type, CNS WHO grade 4
- Histologic diagnosis: glioblastoma
- Grade: 4
- Molecular information: EGFR amplified, negative for IDH1 / IDH2 and histone H3 alterations
Differential diagnosis
- Astrocytoma, IDH mutant:
- Younger individuals, presence of IDH1 / IDH2 gene mutations by immunohistochemistry or sequencing
- Often have ATRX mutation (loss of expression) and p53 mutations (nuclear overexpression)
- Diffuse hemispheric glioma, H3 G34 mutant:
- Older adolescents and young adults (age 11 - 30) with hemispheric mass
- May have classic GBM morphology or primitive neuronal / embryonal morphology
- Characterized by histone H3 G34 mutation on sequencing
- Classically lack Olig2 expression, show ATRX loss of expression (mutant) and have p53 mutations (nuclear overexpression)
- Diffuse midline glioma, H3 K27 altered:
- Midline tumor (brainstem, thalamus, spinal cord, less often basal ganglia or cerebellum)
- Most positive for histone H3K27M mutant protein (nuclear)
- All show loss of histone H3K27 trimethylation (H3K27me3)
- Diffuse pediatric type high grade glioma, H3 wild type and IDH wild type:
- Typically in a child or young adult
- Frequent PDGFRA or EGFR alterations or MYCN amplification
- Methylation profiling may be helpful in difficult cases
- Pleomorphic xanthoastrocytoma:
- Prominent xanthomatous cells
- Often well circumscribed
- Lower grade lesions have no necrosis and low mitotic activity
- Eosinophilic granular bodies (EGBs), Rosenthal fibers and perivascular lymphocytic cuffing
- Often BRAF V600E positive IHC / sequencing and CD34 positive
- Pericellular reticulin deposition
- Lymphoma:
- Tumefactive multiple sclerosis / demyelinating diseases:
- Macrophage rich lesions (CD163 positive) with perivascular lymphoplasmacytic inflammation
- Loss of myelin (LFB) with axonal preservation (neurofilament)
- Creutzfeldt cells: astrocytic cells with nuclear fragmentation may mimic mitotic figures
- Astrocytes have a reactive (fibrillary) appearance, which can be highlighted by GFAP IHC
- Abscess:
- Abundant necrosis with mixed acute and chronic inflammation
- Peripheral granulation tissue and fibrosis
- Neutrophilic inflammation predominates
- Microorganisms may be identified
Additional references
Practice question #1
Which histologic subtype of glioblastoma, IDH wild type (pictured above) is important to diagnose due to increased risk of cerebrospinal fluid (CSF) dissemination?
- Epithelioid
- Gemistocytic
- Primitive neuronal component
- Sarcomatoid
Practice answer #1
C. Primitive neuronal component. Glioblastomas with a primitive neuronal component have an increased risk of CSF spread and potential for distant metastasis in rare case reports. As such, this particular morphologic finding may be important to bring to the attention of the clinical team. A primitive neuronal component in glioblastoma can be diagnosed by identifying a morphologically distinct area with high cellularity, high mitotic activity, loss of expression of glial markers and expression of neuronal markers such as synaptophysin.
Comment Here
Reference: Glioblastoma, IDH wild type
Comment Here
Reference: Glioblastoma, IDH wild type
Practice question #2
BRAF V600E mutations are usually identified in low grade gliomas but are also present in a subset of glioblastomas with which histological features?
- Epithelioid
- Gemistocytic
- Giant cell
- Oligodendrolial-like features
Practice answer #2
A. Epithelioid. BRAF V600E mutations are more frequently encountered in epithelioid glioblastomas and may allow for treatment with targeted therapies (Int J Mol Sci 2018;19:1090).
Comment Here
Reference: Glioblastoma, IDH wild type
Comment Here
Reference: Glioblastoma, IDH wild type
