Adrenal gland & paraganglia
Pheochromocytoma / paraganglioma
Paraganglioma
Authors: Luvy Delfin, M.D., Sylvia L. Asa, M.D., Ph.D.
Editorial Board Member: Bonnie Choy, M.D.
Deputy Editor-in-Chief: Maria Tretiakova, M.D., Ph.D.


Last author update: 18 January 2022
Last staff update: 20 January 2022
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: Paraganglioma[TI] adrenal gland "last 5 years"[DP] review[PT]
Table of Contents
Definition / general | Essential features | Familial pheochromocytoma and paraganglioma syndromes | Terminology | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Gross description | Gross images | Frozen section description | Microscopic (histologic) description | Microscopic (histologic) images | Cytology description | Positive stains | Negative stains | Electron microscopy description | Electron microscopy images | Molecular / cytogenetics description | ICCR guidelines for pheochromocytoma and paraganglioma | AJCC staging | Risk stratification (controversial) | Sample pathology report | Differential diagnosis | Practice question #1 | Practice answer #1 | Practice question #2 | Practice answer #2Cite this page: Delfin L, Asa SL. Paraganglioma. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/adrenalparaganglioma.html. Accessed August 26th, 2025.
Definition / general
- Nonepithelial, neural crest derived neuroendocrine neoplasms arising from the adrenal medulla (pheochromocytoma) and extra-adrenal paraganglia
- Composed of paraneuronal peptide hormone secreting neuroendocrine cells
- Histologically similar, regardless of location
- 2 categories:
- Parasympathetic: arises predominately in the head and neck
- Sympathetic: arises in the retroperitoneum, thorax and pelvis
Essential features
- Nonepithelial neuroendocrine tumors producing dopamine, epinephrine, norepinephrine and other peptide hormones
- Occuring at almost any location, except within the brain and bones
- All tumors have metastatic potential; they are referred to as metastatic or nonmetastatic instead of benign or malignant
- Because of frequency of multifocal primary tumors, metastatic deposits should only routinely be considered as such at sites where normal chromaffin tissue is not present, including bone, brain and lymph node; however, metastatic disease can involve any organ
- Regrowth or recurrence in the surgical bed should not be identified as metastatic pheochromocytoma / paraganglioma
- At least 20% are multiple (bilateral or multicentric)
- Tumor with highest degree of heritability among all human neoplasms
- Hereditary predisposition due to germline mutation present in almost 40% of tumors (see Table 1) (Best Pract Res Clin Endocrinol Metab 2020;34:101416)
Familial pheochromocytoma and paraganglioma syndromes
- Associated with various syndromes and familial conditions, known as familial pheochromocytoma and paraganglioma syndromes, including:
- SDHx related syndromes (SDHB, SDHD, SDHA, SDHC and SDHAF2)
- Von Hippel-Lindau (VHL)
- Neurofibromatosis type 1 (NF1)
- Multiple endocrine neoplasia type 2 (MEN2)
- Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) or FH tumor predisposition syndrome
- TMEM mutations
- MAX mutations
- EPAS1 mutations (Pazak-Zhuang syndrome)
- EGLN1/2 mutations
- MDH2 mutations
- KIF1B mutations
- Multiple endocrine neoplasia type 1 (MEN1)
- Carney triad (gastrointestinal stromal tumor, paraganglioma and pulmonary chondroma): nonhereditary genetic disorder potentially associated with currently unknown somatic mosaicism or SDH genetic defects, including DNA hypermethylation of SDHC or rarely SBHx pathogenic variant
- Genetic testing and counseling should be offered to all patients with paraganglioma regardless of patient and family characteristics (J Intern Med 2019;285:187)
- Genetic status represents a key element for accurate diagnosis, follow up and prognosis
Table 1: susceptibility genes in hereditary pheochromocytoma and paraganglioma
| Gene | Secreting phenotype | Location | Other associated disorders |
| NF1 | Noradrenaline + adrenaline | Adrenal medulla | Multiple neurofibromas, malignant peripheral nerve sheath tumor, brain stem glioma, optic glioma, pilocytic astrocytoma, duodenal neuroendocrine tumors (somatostatinomas), gastrointestinal stromal tumor, café au lait spots, freckling (axilla and groin), Lisch nodules, bone dysplasia |
| VHL | Noradrenaline | Adrenal medulla / abdominal | Retinal and CNS hemangioblastomas, clear cell renal cell carcinoma, renal cysts, pancreatic serous cystadenomas, pancreatic and small bowel neuroendocrine tumors, endolymphatic sac tumor, epididymal papillary cystadenoma, cystadenoma of the broad ligament and mesosalpinx |
| RET | Noradrenaline + adrenaline | Adrenal medulla | Medullary thyroid carcinoma, thyroid C cell hyperplasia, Hirschsprung disease, hyperparathyroidism (multiglandular adenomas), cutaneous lichen amyloidosis, neuromas: lips, tongue, conjunctiva, intestinal ganglioneuromatosis, Marfanoid habitus |
| EPAS1 | Noradrenaline, EPO | Adrenal medulla / thoracic / abdominal | Congenital polycythemia, pancreatic or duodenal somatostatinoma |
| EGLN1 / 2 | Noradrenaline | Adrenal / abdominal / thoracic | Congenital polycythemia |
| SDHA | Noradrenaline, dopamine, nonsecreting | Head and neck / abdominal | Gastrointestinal stromal tumor, renal cell carcinoma, pituitary neuroendocrine tumors |
| SDHB | Noradrenaline, dopamine, nonsecreting | Abdominal / head and neck | Gastrointestinal stromal tumor, renal cell carcinoma, pituitary neuroendocrine tumors |
| SDHC | Noradrenaline, dopamine, nonsecreting | Head and neck | Gastrointestinal stromal tumor, renal cell carcinoma, pulmonary chordoma* |
| SDHD | Noradrenaline, dopamine, nonsecreting | Adrenal, abdominal, head and neck | Gastrointestinal stromal tumor, renal cell carcinoma, pituitary neuroendocrine tumors |
| SDHAF2 | Nonsecreting | Head and neck | ? |
| FH | Noradrenaline, dopamine, nonsecreting | Abdominal | Cutaneous and uterine leiomyomas, renal cell carcinoma |
| TMEM127 | Noradrenaline + adrenaline | Adrenal medulla and abdominal | Renal cell carcinoma |
| MAX | Noradrenaline | Adrenal medulla and abdominal | Parathyroid, pituitary neuroendocrine tumors, pancreatic neuroendocrine tumors, renal cell carcinoma, squamous cell carcinoma, breast, lung and endometrial cancer |
| MDH2 | Noradrenaline, nonsecreting | Head and neck / abdominal | |
| GOT2 | Noradrenaline | Abdominal | |
| SLC25A11 | Noradrenaline, nonsecreting | Abdominal | |
| DLST | Noradrenaline | Thoracic | |
| H3F3A | N/A | Abdominal / head and neck | Giant cell tumor of bone (Clin Cancer Res 2016;22:2301) |
| DNMT3A | Noradrenaline | Head and neck | Papillary thyroid carcinoma, intellectual disability (Cancers (Basel) 2020;12:3304) |
| MET | N/A | Adrenal medulla | Medullary thyroid carcinoma |
| MERTK | N/A | Abdominal | Medullary thyroid carcinoma |
| KIF1B | N/A | Adrenal medulla | Ganglioneuroma / neuroblastoma, leiomyosarcoma, lung adenocarcinoma |
Terminology
- Parasympathetic paraganglioma
- Most commonly arises in the head, neck and upper thorax (along glossopharyngeal and vagus nerve)
- Named according the anatomical sites of origin:
- Carotid body paraganglioma
- Jugulotympanic paraganglioma (previously called glomus jugulare or glomus tympanicum)
- Vagal paraganglioma
- Laryngeal paraganglioma
- Cardiac and pulmonary paraganglioma
- Chemodectoma refers to carotid and aortic body paraganglioma because of their chemoreceptor function but is no longer used
- Often reported to be both biochemically and clinically silent but if measured, frequently produces dopamine
- Sympathetic extra-adrenal paraganglia
- Approximately 85% arise below the diaphragm
- Distributed along prevertebral and paravertebral sympathetic chain and sympathetic nerve fibers innervating retroperitoneum, thorax and pelvis
- Largest paraganglia: adrenal medulla (bilateral) and organ of Zuckerkandl
- Can also arise from cervical sympathetic chain, superior cervical ganglion, ciliary ganglion (orbital paraganglioma)
- Approximately 85% arise below the diaphragm
- Composite paraganglioma:
- Rare
- Combined with ganglioneuroma, ganglioneuroblastoma, neuroblastoma or peripheral nerve sheath tumor
- Most common locations: adrenal, urinary bladder and retroperitoneum
- Misnomers:
- 2 tumors have been called paragangliomas but are not composed of paraganglial chief cells
- They are instead composed of epithelial neuroendocrine cells that are positive for keratins and lack GATA3 expression, often associated with neurons in a gangliocytoma / ganglioneuroma component
- These tumors are not associated with the genetic alterations found in paragangliomas:
- Cauda equina / filum terminale paraganglioma: spinal intradural and extramedullary tumors (3 - 4% of spinal tumors)
- Gangliocytic paraganglioma changing to composite ganglioneuroma neuroendocrine tumor (CoGNET); arises in the duodenal periampular region and has triphasic differentiation: epithelial neuroendocrine cells, spindle shaped Schwannian / sustentacular cells and neuronal ganglion cells (proportion of each cell type varies)
Epidemiology
- Annual incidence of is about 5 cases per million, with 500 to 1,600 cases reported per year in the United States (Eur J Intern Med 2018;51:68)
- Prevalence varies from 0.2% to 0.6% in hypertensive patients to less than 0.05% in the general population (J Hypertens 2020;38:1443)
- Mean age of presentation is 51, plus or minus 16 years
- Distribution by gender is almost equal
- Pediatric paragangliomas are almost always hereditary
- Familial paragangliomas are often multifocal or bilateral
Sites
- Parasympathetic: head and neck, along parasympathetic nerves near carotid body, jugulotympanicum, vagus nerve and larynx
- Carotid body is the most common site
- Sympathetic: 42% adrenal / periadrenal / perirenal, 28% organ of Zuckerkandl / near abdominal aorta, 10% bladder, 12% thorax, 2% cardiac
- Unusual locations: pituitary, paranasal sinuses, orbit, thyroid, parathyroid glands, trachea, heart and lung, posterior mediastinum, gallbladder, liver, gut, pancreas, mesentery and rarely in testes and ovary (J Clin Med 2018;7:280, Am J Surg Pathol 2006;30:600)
Pathophysiology
- Pheochromocytomas and paragangliomas (PPGLs) are neuroendocrine tumors with strong genetic susceptibility, with approximately 40% harboring germline mutation
- Almost 30% of the remaining sporadic cases carry a somatic mutation in a predisposition gene
- Predisposition genes are implicated in mitochondrial metabolism, DNA methylation, chromatin remodeling and mitogen activated protein kinase (MAPK) pathway signaling (see Molecular / cytogenetics description) (Front Mol Neurosci 2019;12:6)
- Co-occurrence of somatic and germline mutations might have a synergistic effect on driver mutations and cosegregation playing a role in tumor progression (Endocr Rev 2017;38:489)
Etiology
- Neural crest derived neoplasm
- Novel experimental evidence suggest that peripheral nerves may represent a stem cell niche for neuroendocrine differentiation in the adrenal medulla and paraganglia
- Peripheral glial stem cells, also called nerve associated Schwann cell precursors (SCPs), have been shown to generate chromaffin cells via an intermediate progenitor (Science 2017;357:eaal3753)
- Role of Schwann cell precursors seems to be less significant in the sympathetic paraganglia around the dorsal aorta (Front Mol Neurosci 2019;12:6)
- Differences in origin of chromaffin cell lineage might explain the variability in biological behavior and genetic landscape related to pheochromocytoma, paraganglioma and neuroblastoma
Clinical features
- Depends on tumor location, size and secretory phenotype (functional versus nonfunctional, sympathetic versus parasympathetic) (see Table 1) (Endocr Rev 2021 Jun 19 [Epub ahead of print])
- Asymptomatic: incidentally detected on imaging studies
- Symptomatic: vary with location and tumor type
- Sympathetic parangliomas: epinephrine, norepinephrine and dopamine
- Adrenal pheochromocytomas and most infradiaphragmatic lesions
- Paroxysmal tachycardia, hypertension, pallor, headache and anxiety
- Micturition induced paroxysms and hematuria caused by urinary bladder paragangliomas
- Mass effect with local compression
- Adrenal pheochromocytomas and most infradiaphragmatic lesions
- Parasympathetic paraganglioma: dopamine or methoxytyramine
- Carotid body:
- Present at the angle of mandible, beneath the anterior edge of the sternocleidomastoid muscle, lateral to the tip of the hyoid bone (bifurcation of common carotid artery)
- Usually slow growing, painless mass; often pulsatile
- More frequent in patients living at high altitudes and with history of chronic hypoxia
- May cause cranial nerve palsy, dysphagia, hoarseness or carotid sinus syndrome (bradycardia and syncope)
- Mainly secrete dopamine
- May adhere to carotid bifurcation and involve vagus nerve (group II and III Shamblin classification)
- May invade locally or metastasize to lymph nodes or to the lungs (must rule out multicentric synchronous or metachronous tumor)
- Jugulotympanic:
- Usually arise lateral in temporal bone or jugular bulb, erode through floor and present as mass of external auditory canal or middle ear; also mass at base of skull, middle ear polyps
- 40% extend into cranial cavity
- Usually adults; female preponderance
- Often misidentified as hemangiomas
- Bone involvement can be present
- Vagal / intravagal:
- Arise from small dispersed collection of paraganglia following the cervical course of the vagus nerve (jugular and nodal ganglia)
- Symptoms: painless neck mass, hoarseness, dysphagia, weakness of the tongue, vocal cord paralysis and Horner syndrome
- More frequent on the right side
- More common in women
- Mean age: 50 years
- Laryngeal:
- Rare
- Usually arise from superior paraganglia
- Hoarseness and dysphagia
- 25% mortality
- Often tender subcutaneous metastases
- Aorticopulmonary:
- May cause hoarseness, dysphagia or chest pain / discomfort
- Rarely hemoptysis or superior vena cava syndrome
- Carotid body:
- Composite paraganglioma:
- Extremely rare tumors
- Age at diagnosis: 15 months to 81 years
- Slight female predominance
- Most common paraganglioma with ganglioneuroma
- Most common location: adrenal, urinary bladder and retroperitoneum
- Clinically silent, associated with catecholamies related signs or rarely associated with watery diarrhea, hypokalaemia and achlorhydria (WDHA syndrome) due to secretion of vasoactive intestinal polypeptide (VIP)
Diagnosis
- Distinct signs and symptoms on presentation (as described above) (Pancreas 2021;50:469)
- Increase levels of fractionated metanephrines and catecholamines in plasma and urine (values 3 - 4 times higher than the upper reference limit are almost always considered diagnostic for PPGLs)
- Anatomical documentation of the tumor by imaging studies
- Sometimes asymptomatic and discovered incidentally
Laboratory
- Plasma and urinary metanephrines or catecholamines levels (liquid chromatography tandem mass spectrometry based) (Pancreas 2021;50:469)
- PPGLs can be classified according to their biochemical profile (N Engl J Med 2019;381:552)
- Truly biochemically silent phenotype (usually head and neck region)
- Biochemically pseudosilent phenotype: normal or near normal levels of catecholamines and metanephrines
- Noradrenergic phenotype: increased levels of norepinephrine NE / NMN (usually extra-adrenal)
- Adrenergic phenotype: either purely elevated epinephrine (E) / metanephrine (MN) or in both E / MN and NE / NMN (usually adrenals)
- Dopaminergic phenotype: high levels of DA / 3-methoxytyramine (3-MT) with normal or near normal levels of E / MN and NE / NMN (extra-adrenal and mainly in the head and neck region
- Plasma chromogranin A levels as a complement to metanephrines assays can be used as tumor marker to follow disease progression (Pancreas 2021;50:469)
- PPGLs can be classified according to their biochemical profile (N Engl J Med 2019;381:552)
Radiology description
- Anatomical imaging studies: Doppler ultrasonography, CT, MRI, MRA, CT angiography (Cancer Imaging 2012;12:153, Insights Imaging 2019;10:29, Eur J Nucl Med Mol Imaging 2019;46:2112)
- CT is the anatomic imaging modality of choice due to its excellent spatial resolution
- MRI is recommended for children, pregnant women or patients with hereditary syndromes or metastatic disease
- Noncontrast CT: soft tissue density
- Contrast enhanced CT: well circumscribed, lobulated heterogeneously enhancing soft tissue density mass (J Radiol Case Rep 2019;13:19)
- Contrast enhanced MRA: early and strong contrast enhancement of hypervascular mass in a typical localization with intense tumor blush in the arterial phase (AJNR Am J Neuroradiol 2008;29:883)
- MRI: lobulated oval well defined lesion mass, signal intensity similar to muscle on T1 weighted and increased signal on T2 weighted, with well defined flow voids on T2 weighted images (Acta Univ Palacki Olomuc Fac Med 1998;141:27, Neuroradiology 2021;63:547)
- Functional imaging studies: there are 3 types of PET / CT radiopharmaceuticals that exert their actions through different receptors: 18F-FDG, 18F-fluorodopa (18F-FDOPA) and 68Galium(68Ga)-tetraazacyclododecanetetraacetic acid (DOTA) analogs (Front Endocrinol (Lausanne) 2018;9:515)
- Ga 68-DOTATATE PET / CT is the preferred modality, binds to somatostatin receptor (SSTR2) expressed in paragangliomas with a detection rate of 80 - 100%, demonstrating increased uptake in the tumor anatomical location
- See Radiology images
Radiology images

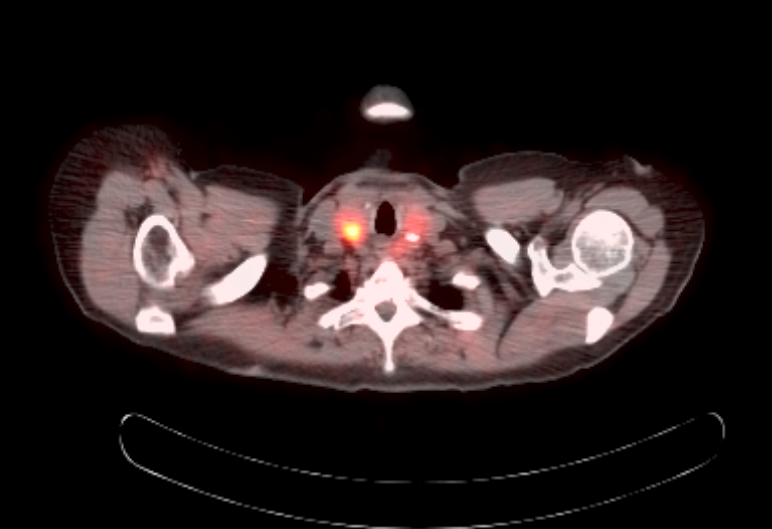
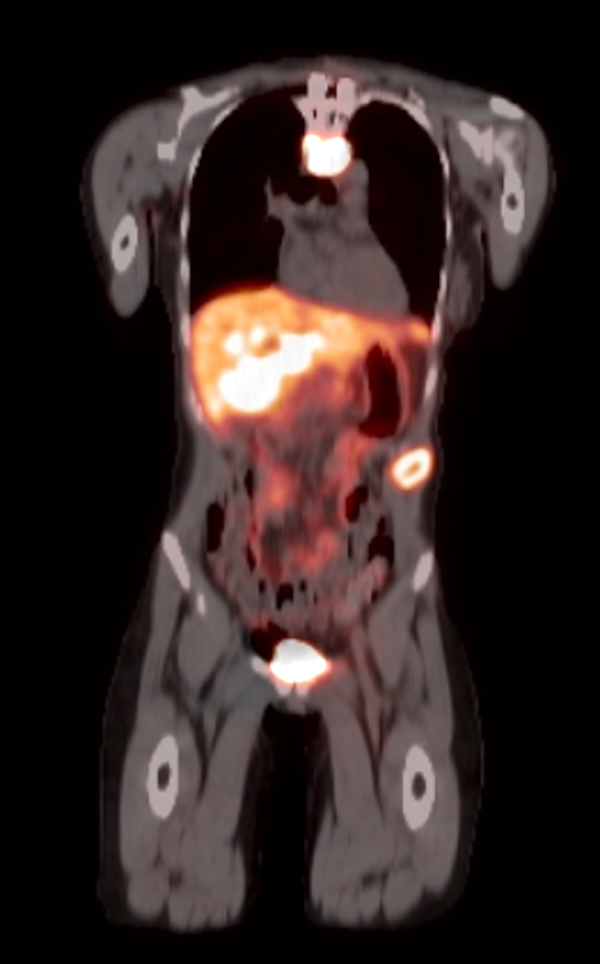


Prognostic factors
- Aggressive biologic behavior can be due to metastases or locally aggressive / multicentric inoperable disease (Pancreas 2021;50:469)
- Poor clinical outcome is also related to high Ki67 labelling index
- In addition, low tumoral and plasma levels of chromogranin B (CHGB) have recently been suggested as a marker for potential aggressive behavior (Am J Surg Pathol 2019;43:409)
- Molecular predictive and prognostic markers:
- Several markers with different levels of evidence have been proposed as genetic and expressional indicators of metastatic disease (Cancers (Basel) 2019;11:225)
- Presence of constitutional SDHB mutations is the strongest genetic risk factor for the development of metastasis
- Other significant markers include somatic mutations in ATRX or SETD2
- High somatic mutational burden and global hypermethylation status
- TERT gene abberancies
- Gene fusions involving MAML3
- 2 mRNA clustering subtypes: the pseudohypoxia (cluster 1) and the Wnt altered (cluster 2)
Case reports
- Paraganglioma:
- 8 year old boy presenting with stridor caused by tracheal SDHC related hereditary paraganglioma (Int J Pediatr Otorhinolaryngol 2018;107:145)
- 15 year old girl with irregular menses and polypoid vaginal SDHB related hereditary paraganglioma (Int J Gynecol Pathol 2020;39:599)
- 24 year old man with recurrent jugulotympanic paraganglioma showing progressive loss of sustentacular cells (Endocr Pathol 2020;31:310)
- 24 year old man with primary paraganglioma of the left renal pelvis (Tumori 2017;103:e47)
- 26 year old man presenting with cardiac arrest during physical activity due to primary cardiac paraganglioma with germline SDHB mutation (J Surg Oncol 2018;117:160)
- 30 year old man with succinate dehydrogenase B gene mutation and left atrial and carotid body paraganglioma (Ann Thorac Surg 2017;103:e323)
- 41 year old man presenting with a clinically functional cardiac paraganglioma and rare SDHC mutation (Endocr Pathol 2014;25:315)
- 47 year old woman with both a pheochromocytoma and a multilocular clear cell RCC driven by a novel germline mutation in the TMEM127 gene (Virchows Arch 2015;466:727)
- 49 year old woman with primary hepatic functional paraganglioma (Medicine (Baltimore) 2018;97:e0293)
- 53 year old man presenting with hematuria, hypertension and headache due to bladder paraganglioma (Pan Afr Med J 2020;36:339)
- 54 year old woman with large paraganglioma in left paraaortic area (Asian Cardiovasc Thorac Ann 2012;20:482)
- 61 year old postmenopausal woman with ovarian carcinoma and paraaortic paraganglioma mimicking lymph node metastasis (J Obstet Gynaecol Res 2010;36:204)
- 68 year old woman with retroperitoneal tumor and liver metastases 23 years after resection (Arch Pathol Lab Med 1985;109:373)
- 73 year old woman with primary sellar paraganglioma (World Neurosurg 2019;125:32)
- 75 year old woman with paraganglioma of orbit and regression (Orbit 2015;34:99)
- 76 year old man with mesenteric paraganglioma (Arch Pathol Lab Med 2002;126:362)
- 82 year old woman with gastric paraganglioma (G Chir 2017;38:84)
- Pheochromocytoma:
- 7 year old boy presenting with cardiomyopathy due to left sided pheochromocytoma (BMC Pediatr 2020;20:299)
- 14 year old child with recurrent sporadic malignant pheochromocytoma with retroperitoneal lymphadenopathy (J Indian Assoc Pediatr Surg 2017;22:242)
- 15 year old woman presenting with ectopic Cushing syndrome caused by pheochromocytoma (Chin Med J (Engl) 2012;125:1193)
- 27 year old man presenting with reversible posterior leukoencephalopathy syndrome related to pheochromocytoma (Medicine (Baltimore) 2020;99:e20918)
- 52 year old man presenting with brain metastasis from pheochromocytoma (Brain Tumor Res Treat 2018;6:101)
- 61 year old woman presenting with an incidental large cystic pheochromocytoma (Scott Med J 2020;65:64)
- Composite paraganglioma / phechromocytoma:
- 42 year old man presenting with spontaneous rupture of a composite pheochromocytoma with malignant peripheral nerve sheat tumor (Asian J Surg 2016;39:187)
- 45 year old man presenting with dysuria and flank pain caused by a composite paraganglioma with neuroblastoma of the bladder (Curr Urol 2015;8:208)
- 47 year old man presented with low back pain and tingling cuased by a composite paraganglioma with ganglioneuromatous elements at the filum terminale (J Neurosurg Spine 2010;12:709)
- 50 year old woman presenting with a composite paraganglioma in the soft tissue adjacent to the carortid artery (Int J Surg Pathol 2019;27:282)
- 59 year old man presenting with a composite paraganglioma-ganglioneuroma concomitant with adrenal metastasis of medullary thyroid carcinoma in a patient with MEN2B (Asian J Endosc Surg 2017;10:66)
Treatment
- Complete surgical excision is the primary treatment (North American Neuroendocrine Tumor Society guidelines April 2021) (Pancreas 2021;50:469)
- Intraoperative risk is kept to the minimum with appropriate preoperative medical treatment:
- Blockage of catecholamine effect for 10 - 14 before surgery with alpha adrenoreceptor blockade followed by beta adrenoreceptor blockade
- Preoperative volume expansion with saline infusion and increase water intake to limit volume contraction after surgery
- Laparoscopic surgery is the first option for adrenal and extra-adrenal tumors
- Partial cortical sparing adrenalectomies are advocated for bilateral adrenal disease
- Long term periodic follow up is always recommended
- Plasma free or fractionated urine metanephrines
- Recommended at 6 and 12 months following resection (every 3 - 6 months for advanced disease), then annually
- Duration of follow up not defined
- Chromogranin A (neuroendocrine marker): consider if tumor does not produce plasma metanephrines
- Advanced disease management:
- Palliative surgery
- Radiofrequency ablation or cryoablation of metastatic lesions
- High dose versus fractionated 131I MIBG is used in patients with positive 123-I MIBG scintigraphy
- Chemotherapy provide tumor regression and symptom relief in up to 50% of patients with negative 123-I MIBG scintigraphy
- External beam irradiation represent and appropriate approach for bone metastasis
Gross description
- Firm, well circumscribed to infiltrative and partially encapsulated or pseudoencapsulated (Mariani-Costantini: Carotid Body and Vagal Paragangliomas - Epidemiology, Genetics, Clinicopathological Features, Imaging, and Surgical Management [Accessed 4 November 2021])
- Varies from dusky brown to red, gray or tan cut surface (J Radiol Case Rep 2019;13:19)
- Hemorrhage and cystic degeneration may be present
Gross images
Contributed by Luvy Delfin, M.D., Sylvia L. Asa, M.D., Ph.D. and Debra Zynger, M.D.
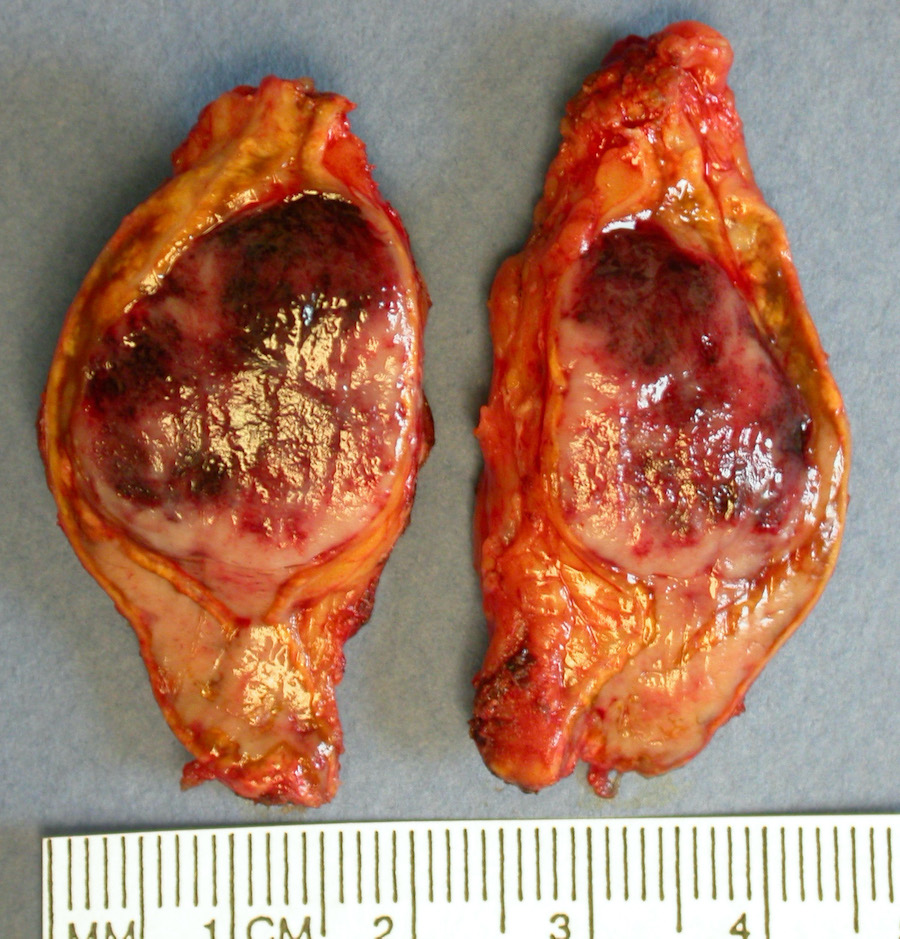
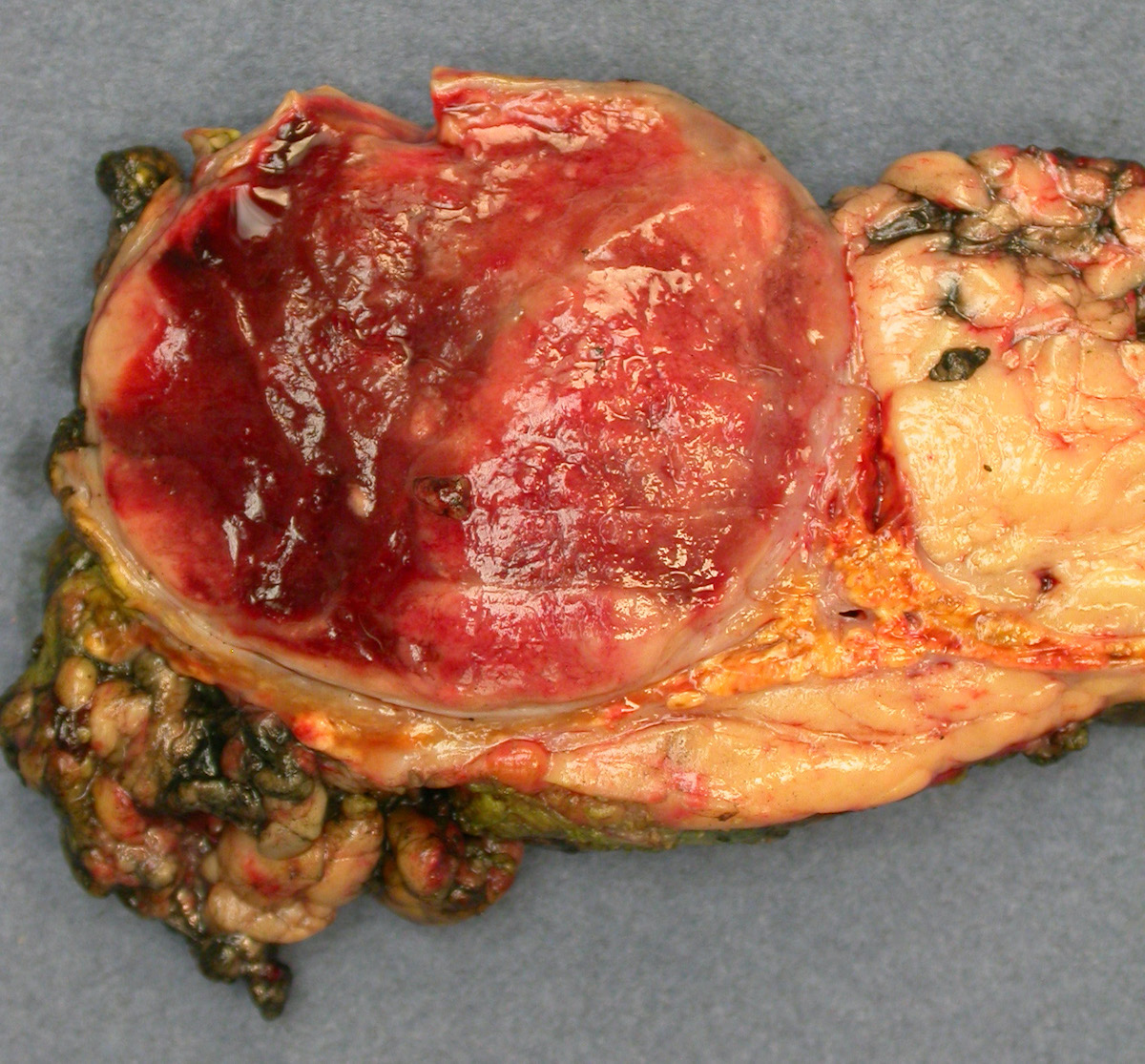
Adrenal pheochromocytoma
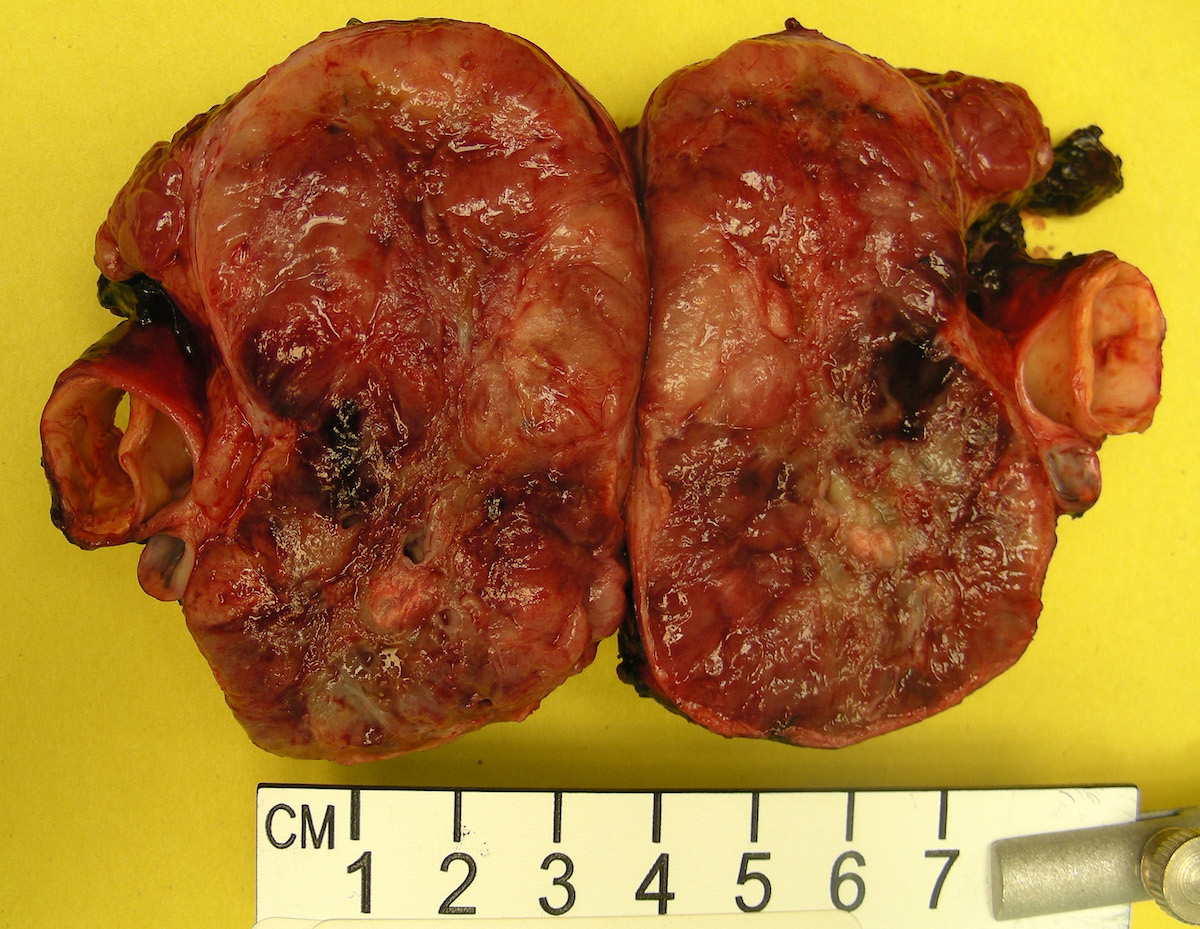
Para-aortic paraganglioma
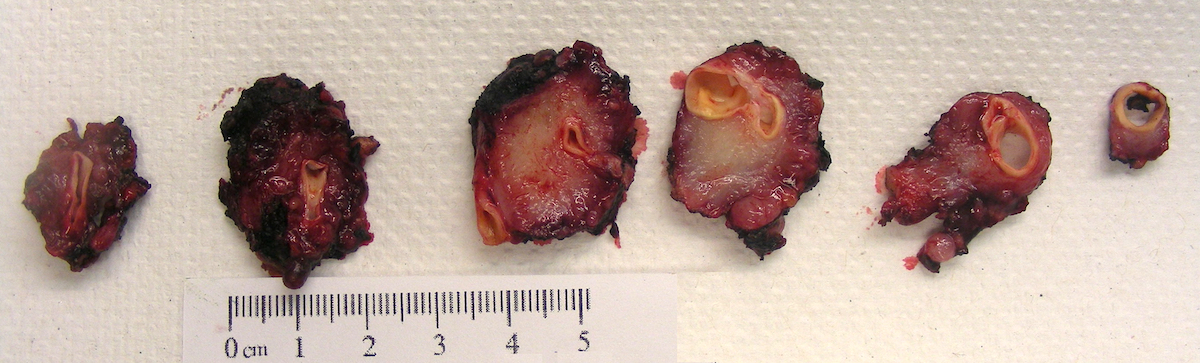
Carotid body paraganglioma
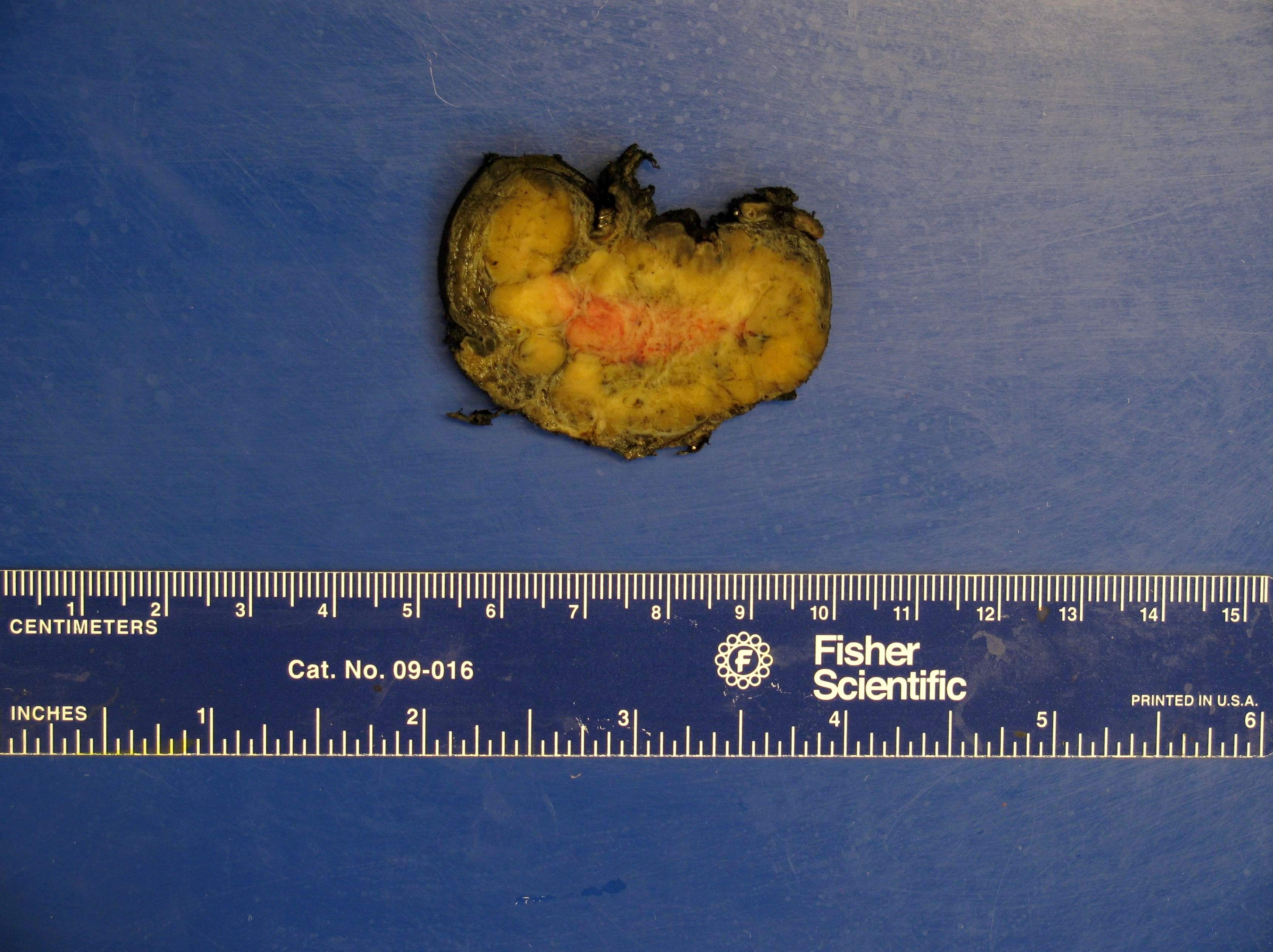
Parapharyngeal paraganglioma
Frozen section description
- Intraoperative consultation is rarely requested and generally discouraged
- Correct frozen diagnosis can be extremely challenging if low clinical suspicion and unusual location (Int J Surg Pathol 2018;26:213)
- Tumors are often misdiagnosed due to their dissimilar architectural patterns and cytologic features worsened by freezing artifact
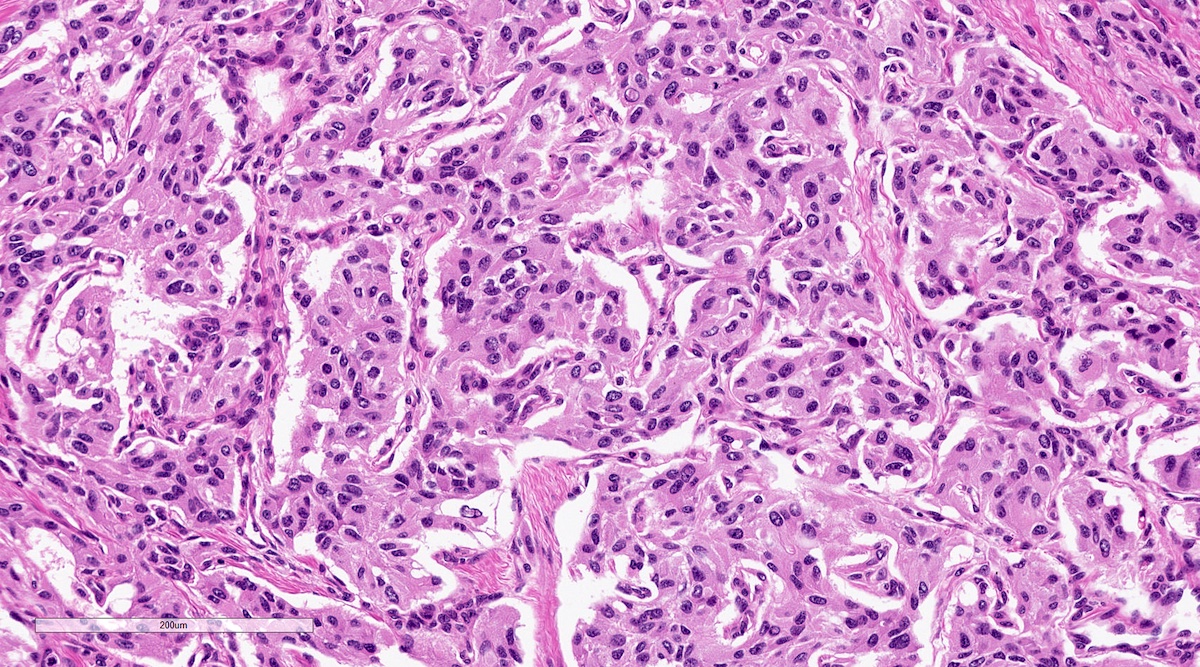
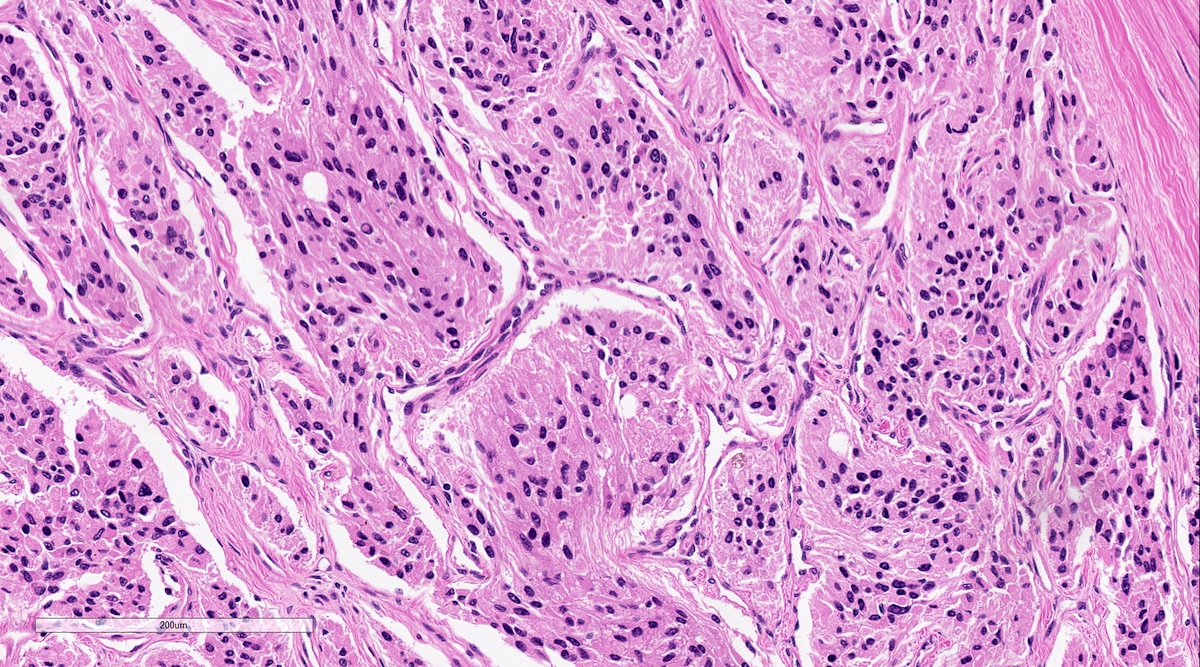
Microscopic (histologic) description
- Prevailing histologic pattern: epithelioid chief cells arranged in distinctive clusters / nests (zellballen pattern), separated by prominent fibrovascular stroma (J Clin Med 2018;7:280)
- Trabecular pattern: ribbons or cords of epithelioid cells divided by fibrous bands
- Other patterns: pseudorosette, angioma-like, spindled and sclerosing
- Chief cells: round, oval to polygonal cells with abundant granular basophilic, eosinophilic or amphophilic cytoplasm (Surg Pathol Clin 2019;12:951)
- Intracytoplasmic hyaline globules may be present in sympathoadrenal paragangliomas
- Giant multinucleated cells and bizarre cells can be present (Srp Arh Celok Lek 2002;130:7)
- Rarely, elongated and spindle shaped cells with a sarcomatoid appearance may be found
- Scattered ganglion cells can be seen
- May have nuclear atypia
- May have dysmorphic vessels, melanin-like pigment (neuromelanin) (pigmented paraganglioma), amyloid, abundant stroma and osseous metaplasia (Diagn Pathol 2012;7:77, Hum Pathol 1992;23:33)
- No or rare mitotic figures except in highly aggressive rapidly proliferating lesions
- May have focal chronic inflammatory infiltrate
- Necrosis is unusual except in patients who have undergone preoperative tumor embolization
- Special histopathologic features usually related to genetic syndromes:
- VHL syndrome: prominent stromal edema, clear cytoplasm and lipid degeneration (Am J Surg Pathol 1987;11:480)
- SDHx related syndrome: granular eosinophilic cytoplasm (Am J Surg Pathol 2020;44:422)
- MEN2 syndrome: unilateral or bilateral adrenal medullary hyperplasia (Neoplasia 2014;16:868)
Microscopic (histologic) images
Contributed by Luvy Delfin, M.D. and Sylvia L. Asa, M.D., Ph.D.
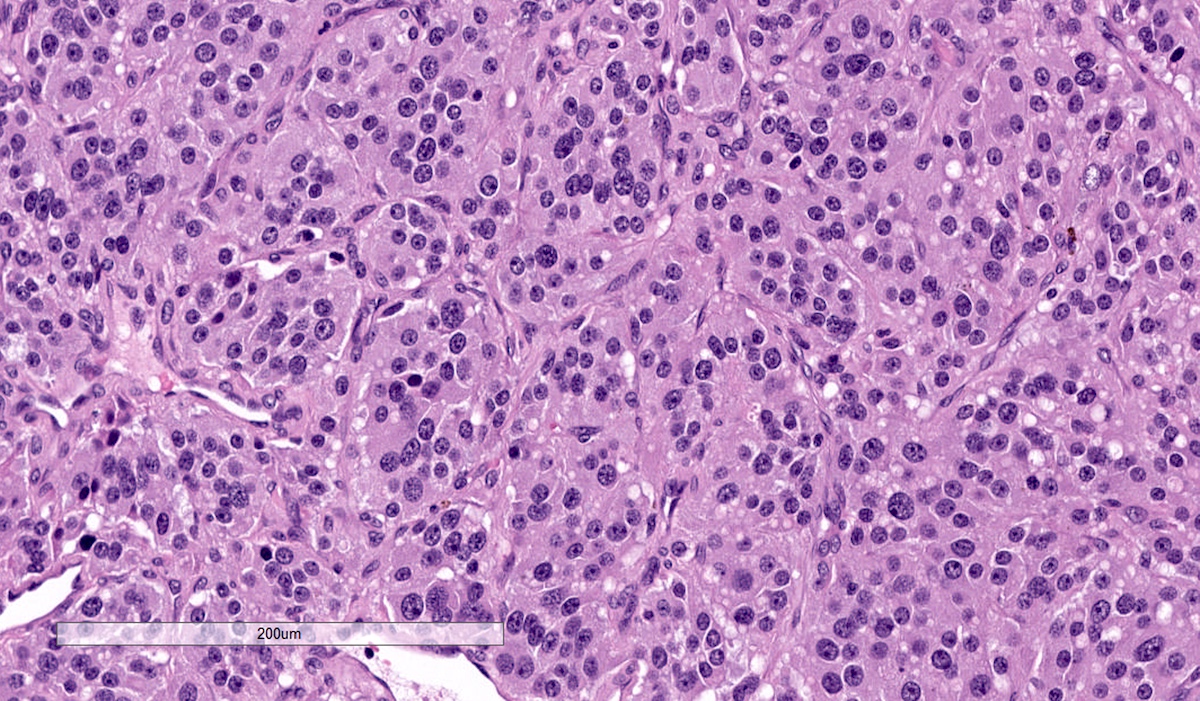
Adrenal pheochromocytoma
Duodenum
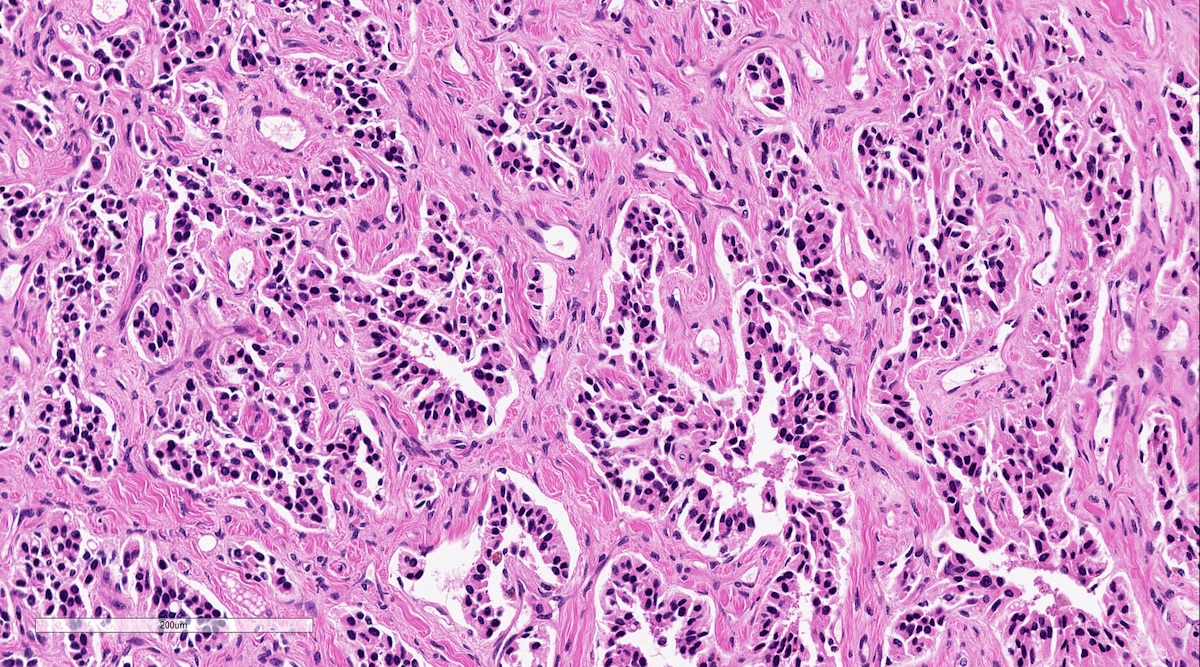
Pheochromocytoma
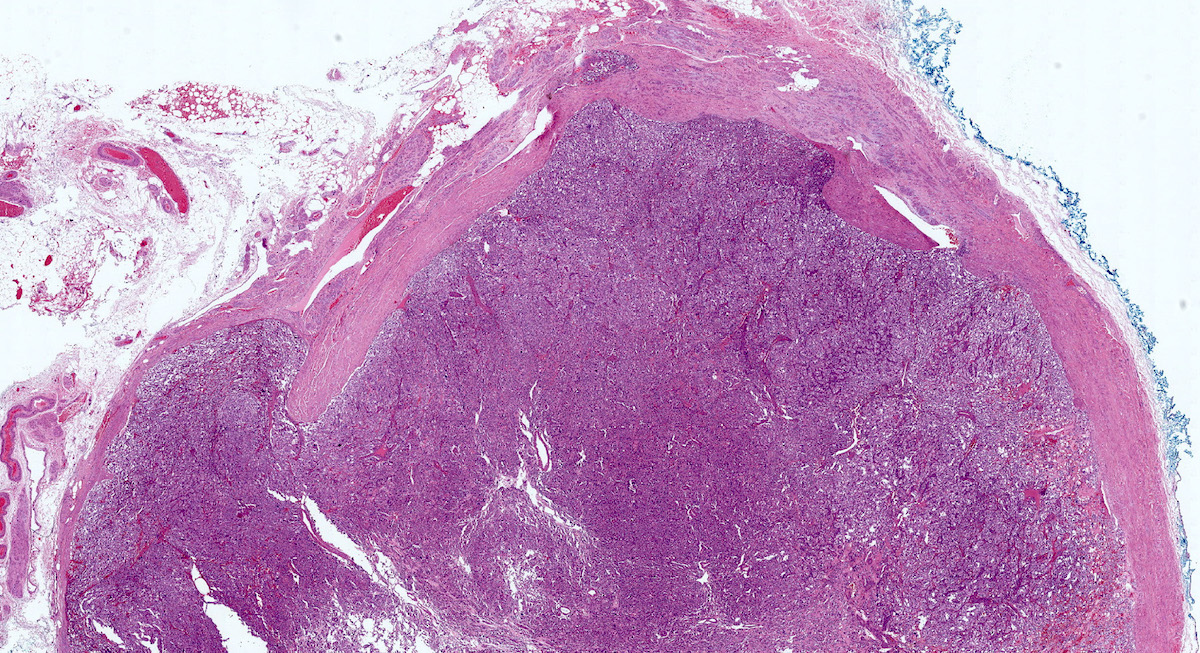
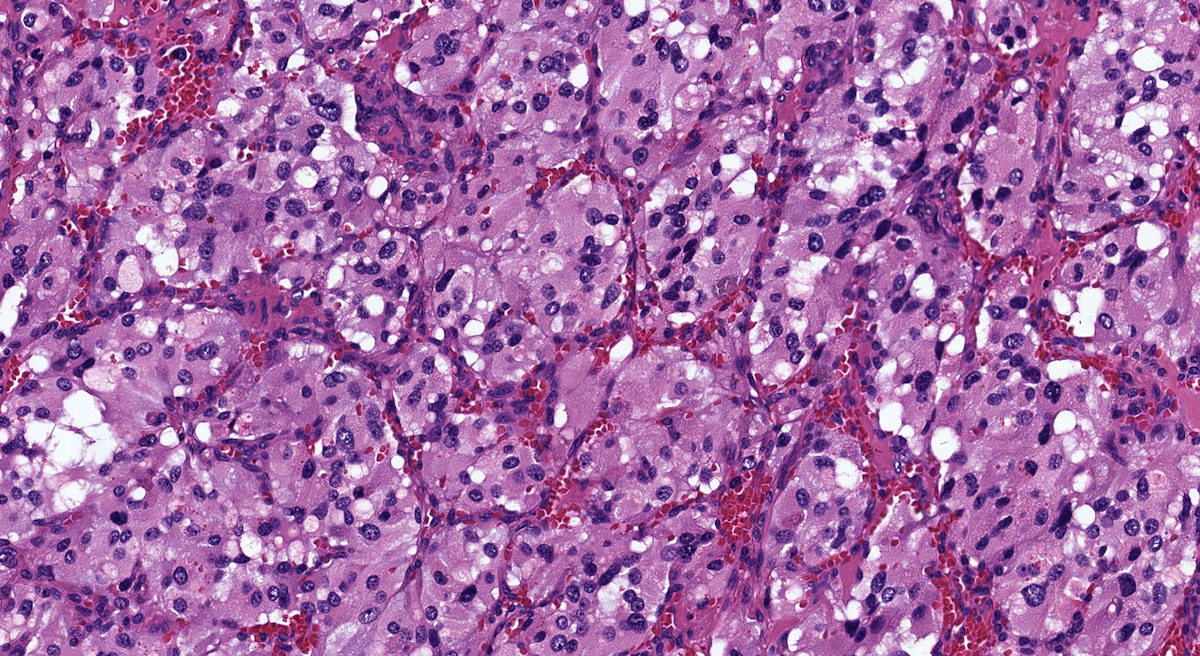
Organ of Zuckerkandl
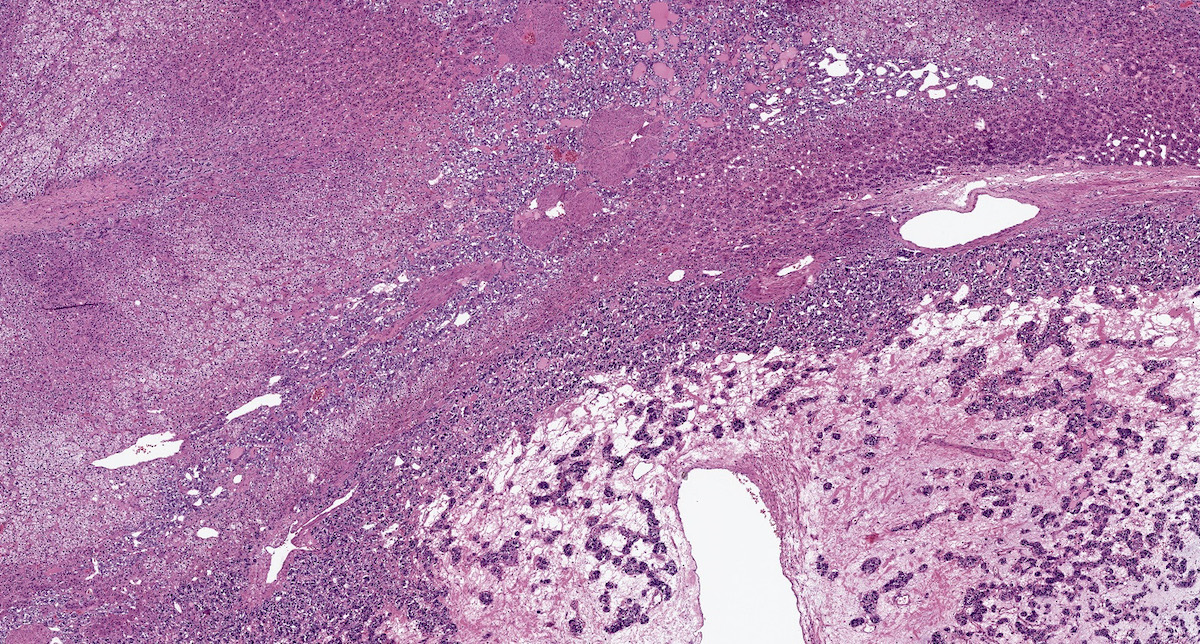
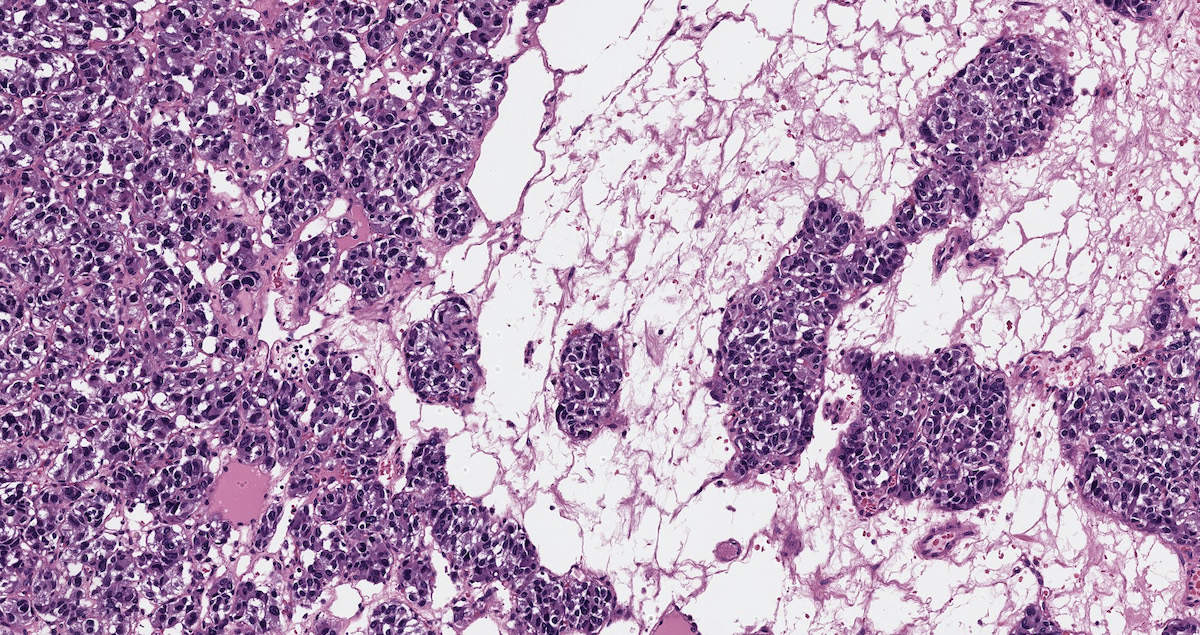
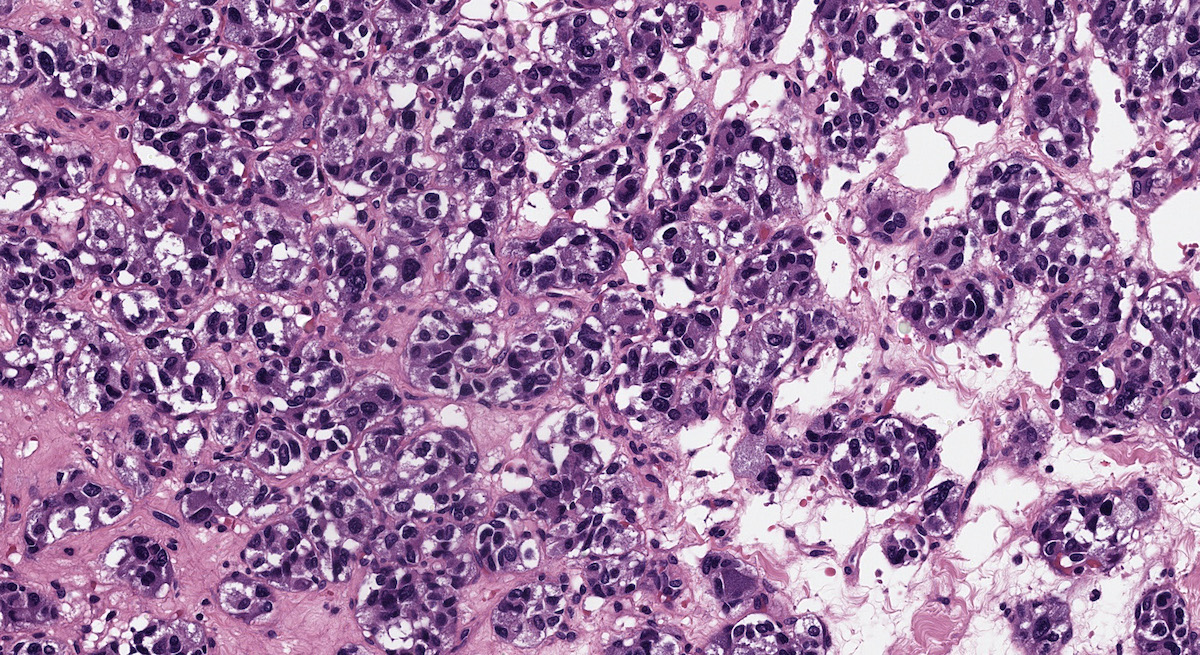
Pheochromocytoma in VHL
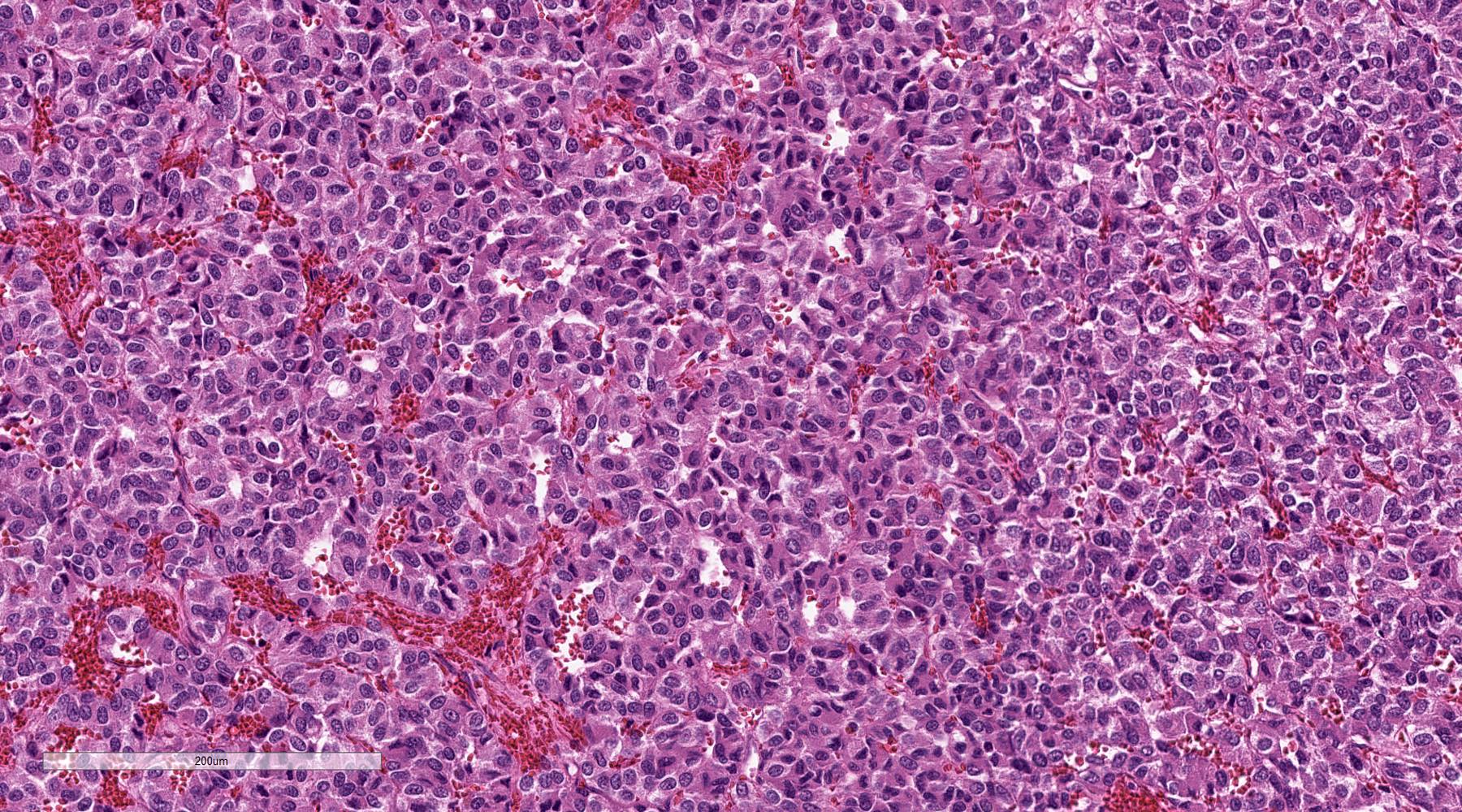
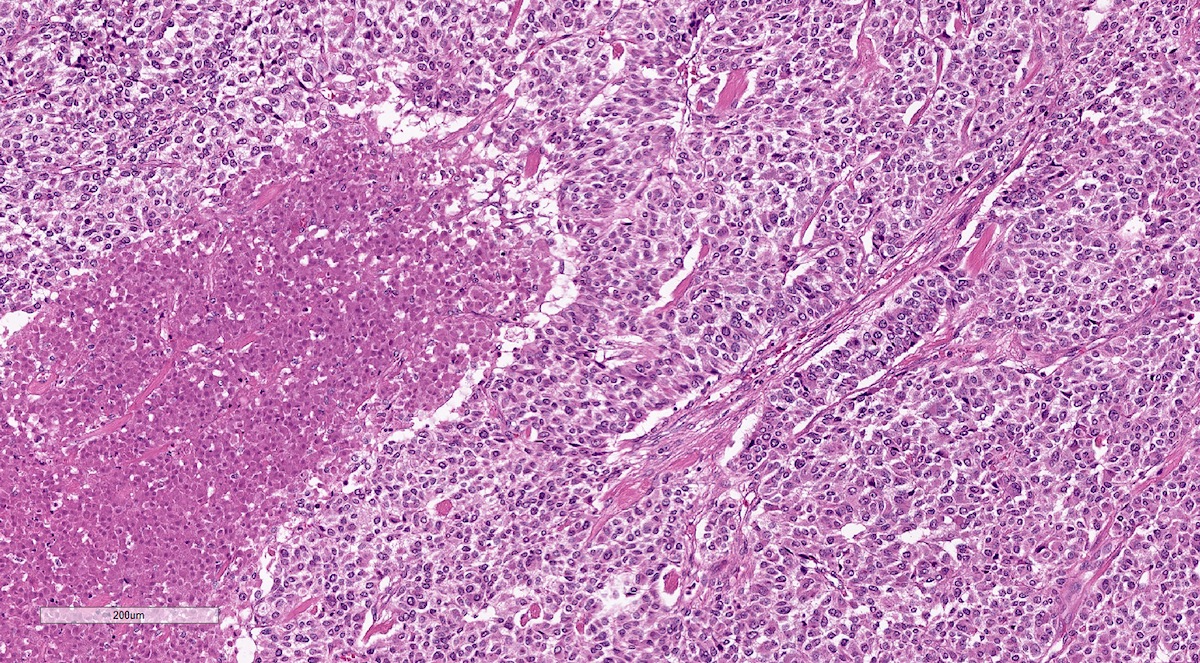
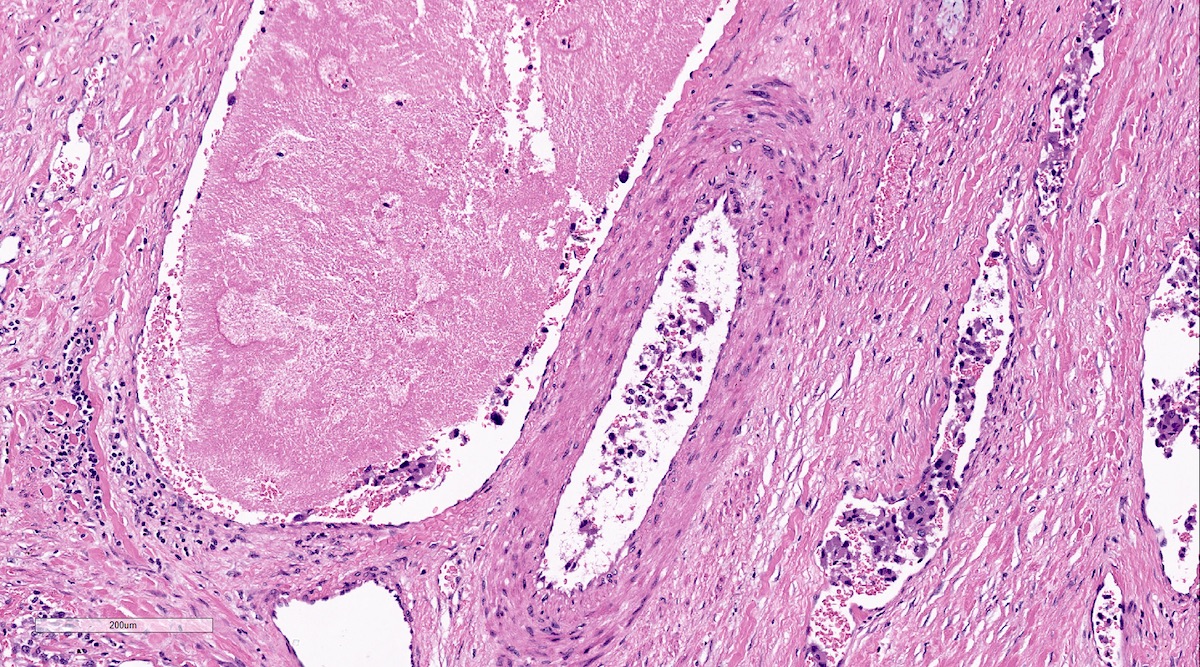
Pheochromocytoma with metastatic behavior
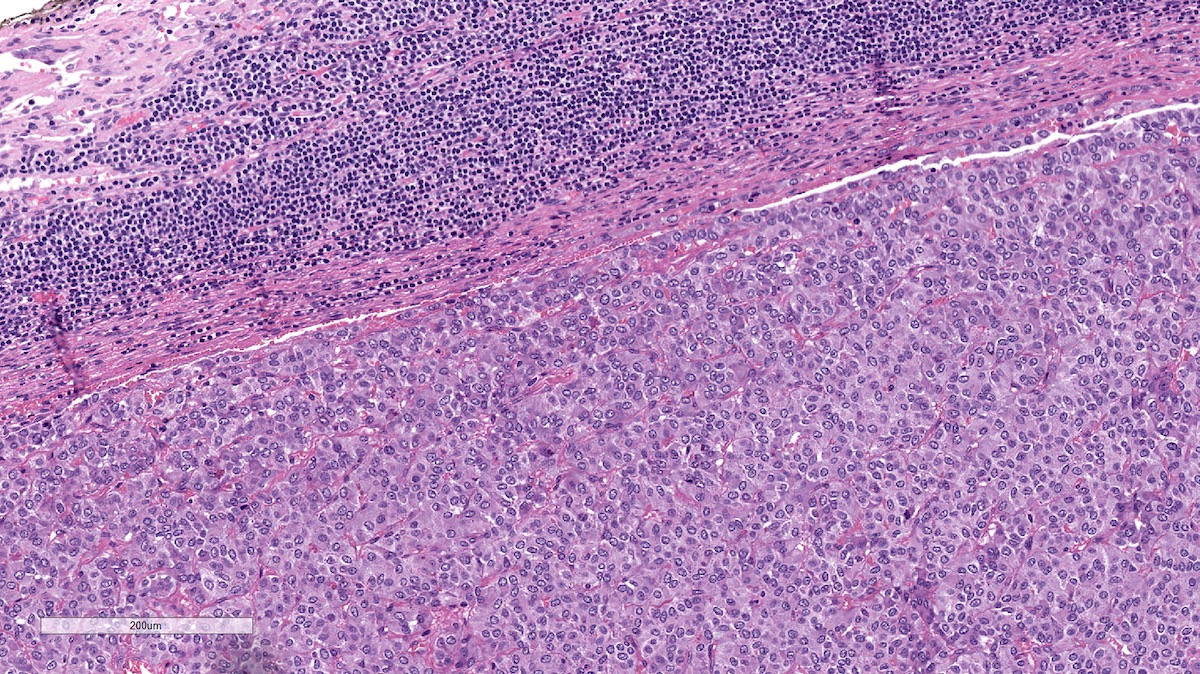
Metastatic pheochromocytoma in lymph node
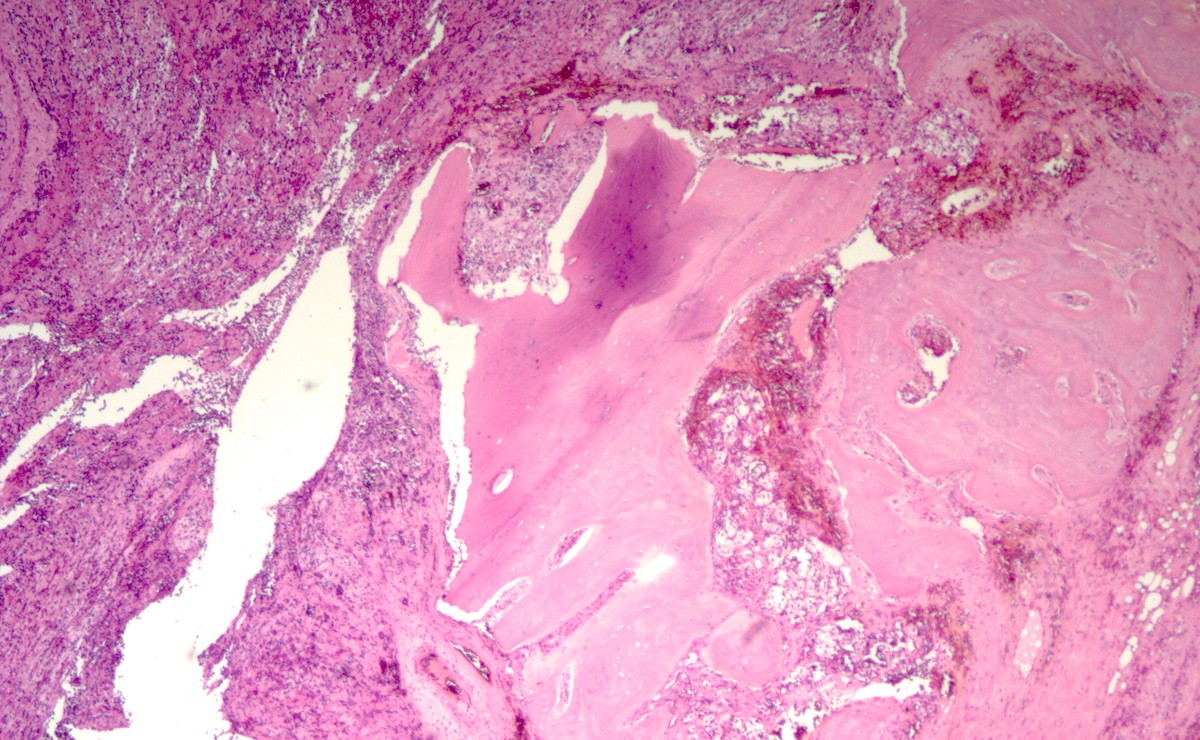
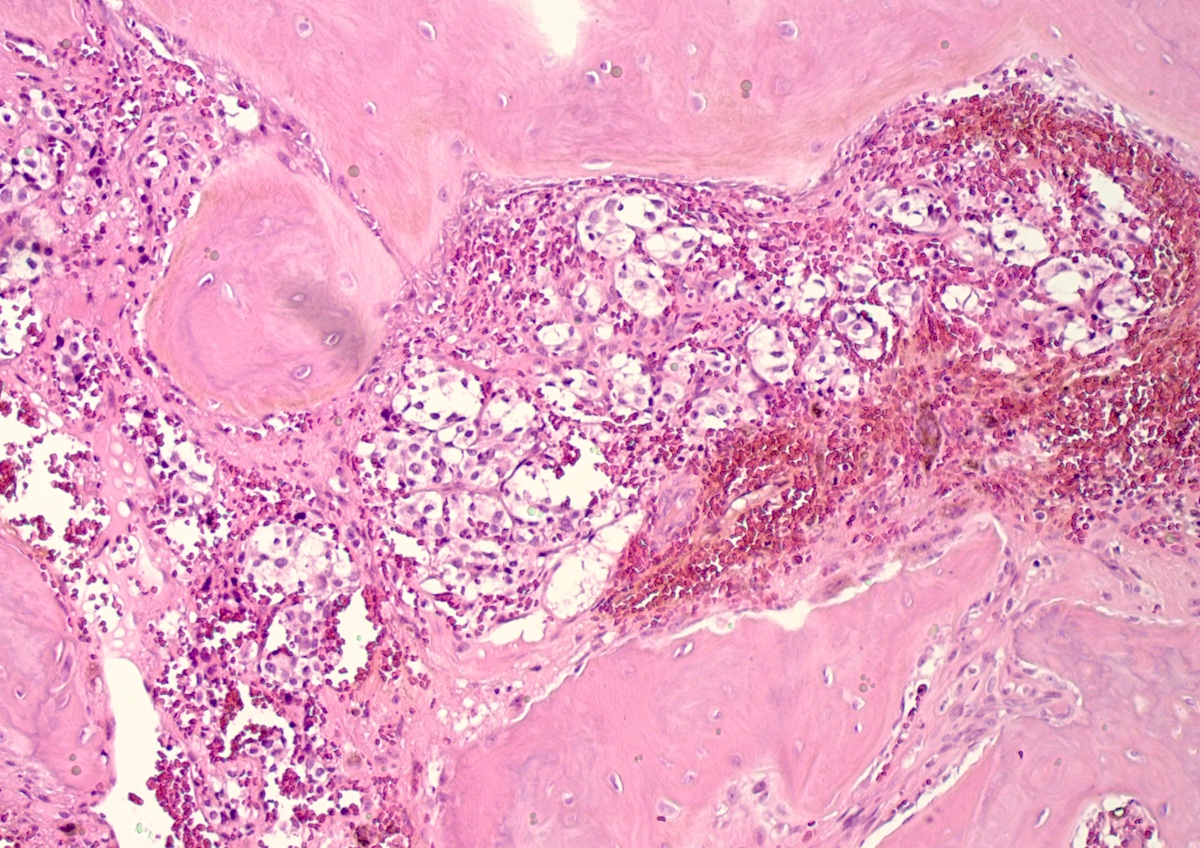
Metastatic paraganglioma in bone
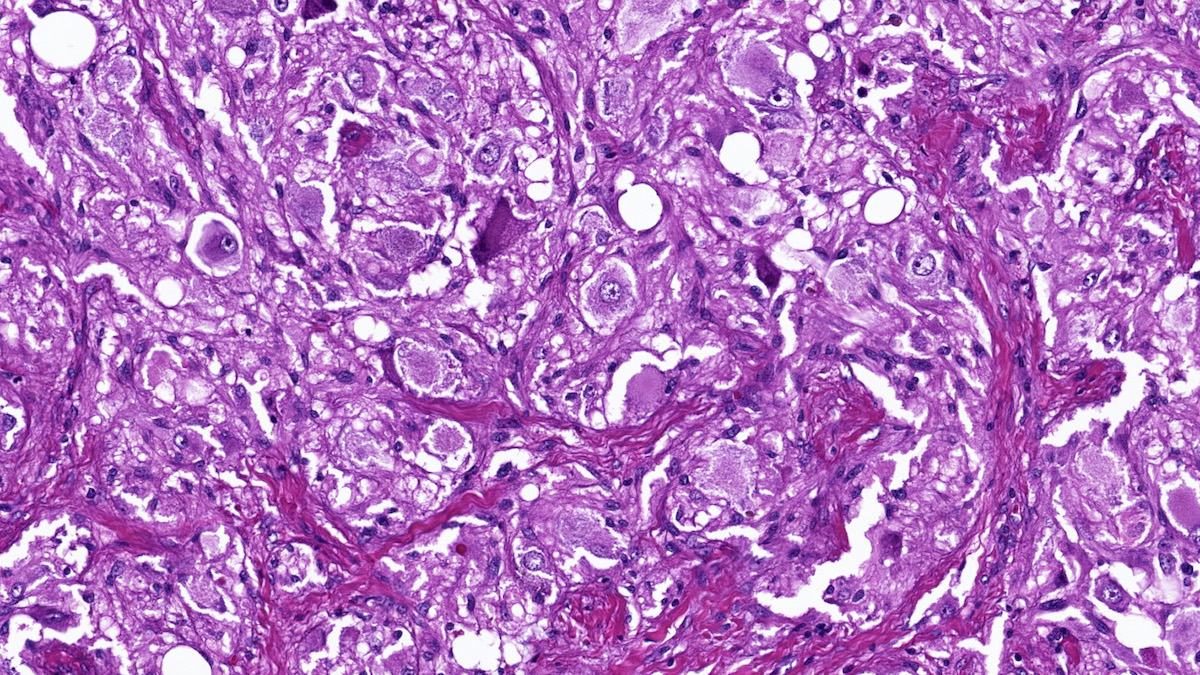
Composite pheochromo-
cytoma with ganglioneuroma
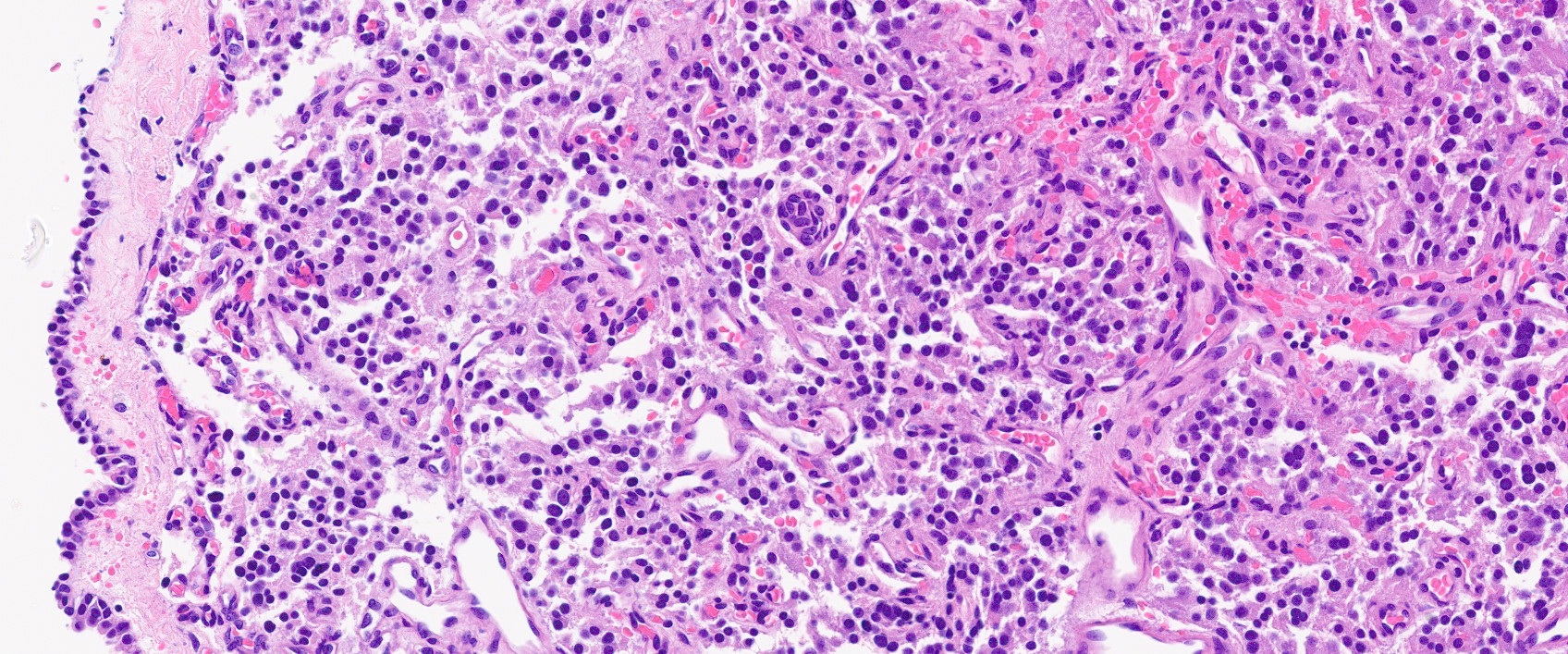
Middle ear: jugulotympanic paraganglioma
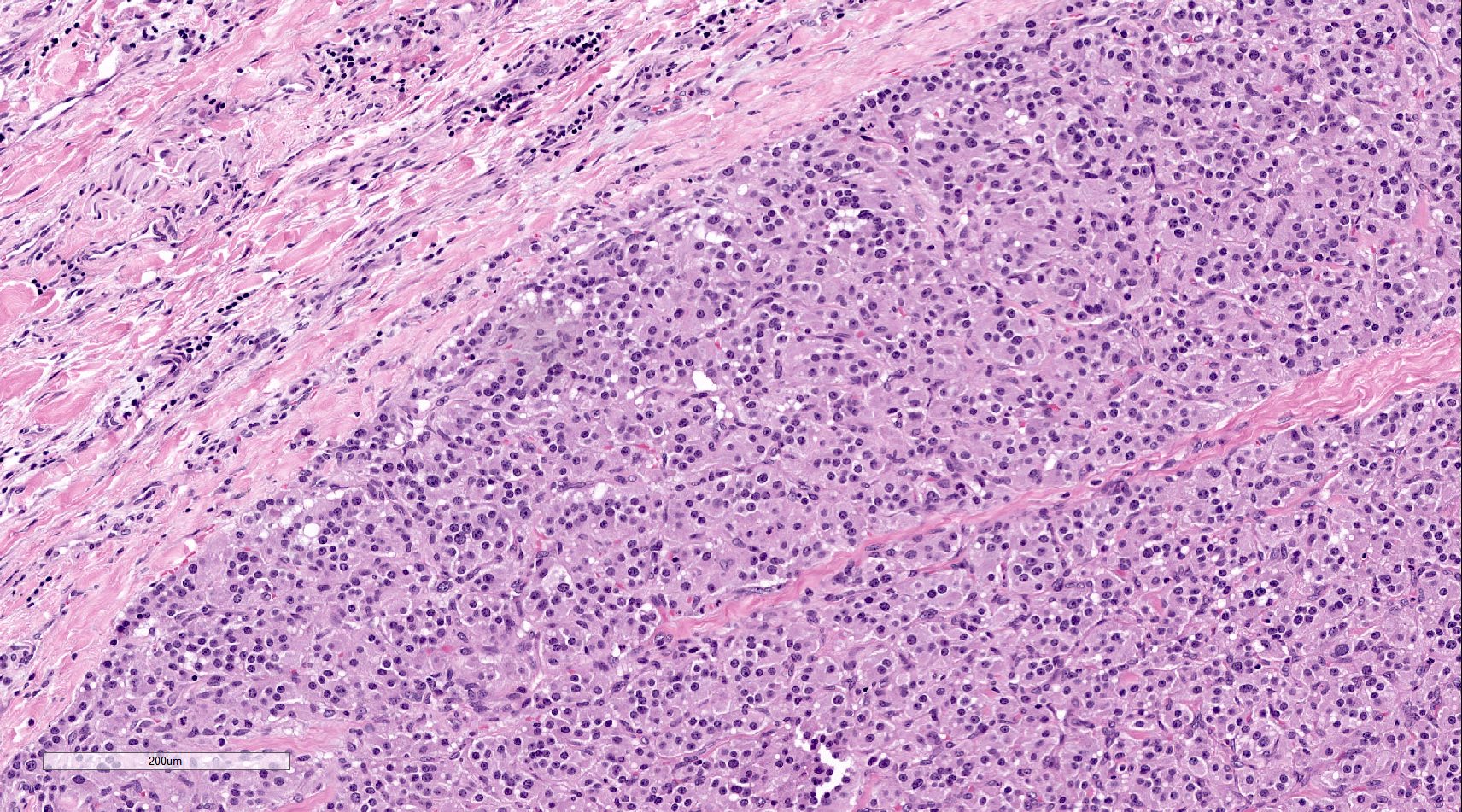
Cardiac paraganglioma
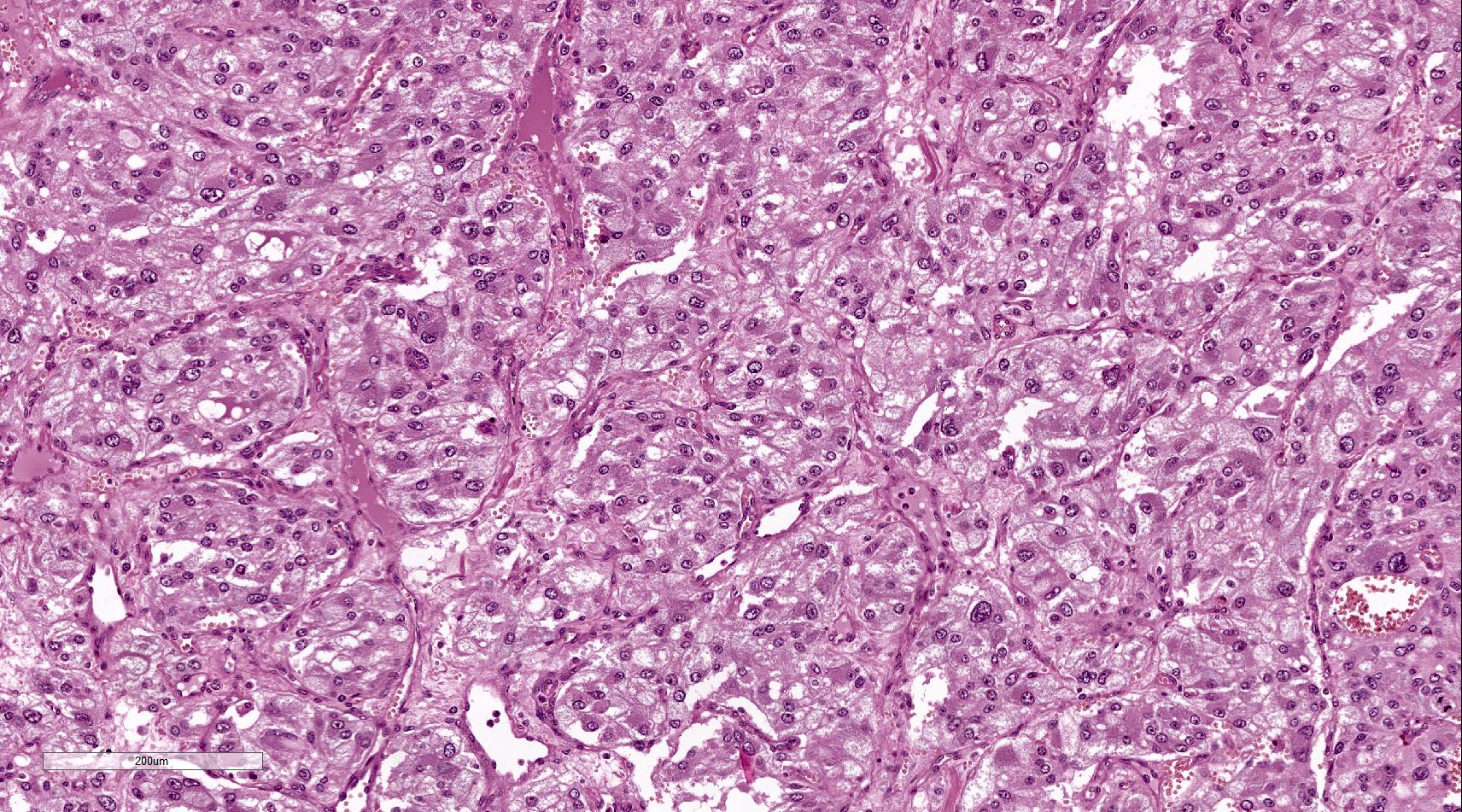
Para-aortic paraganglioma
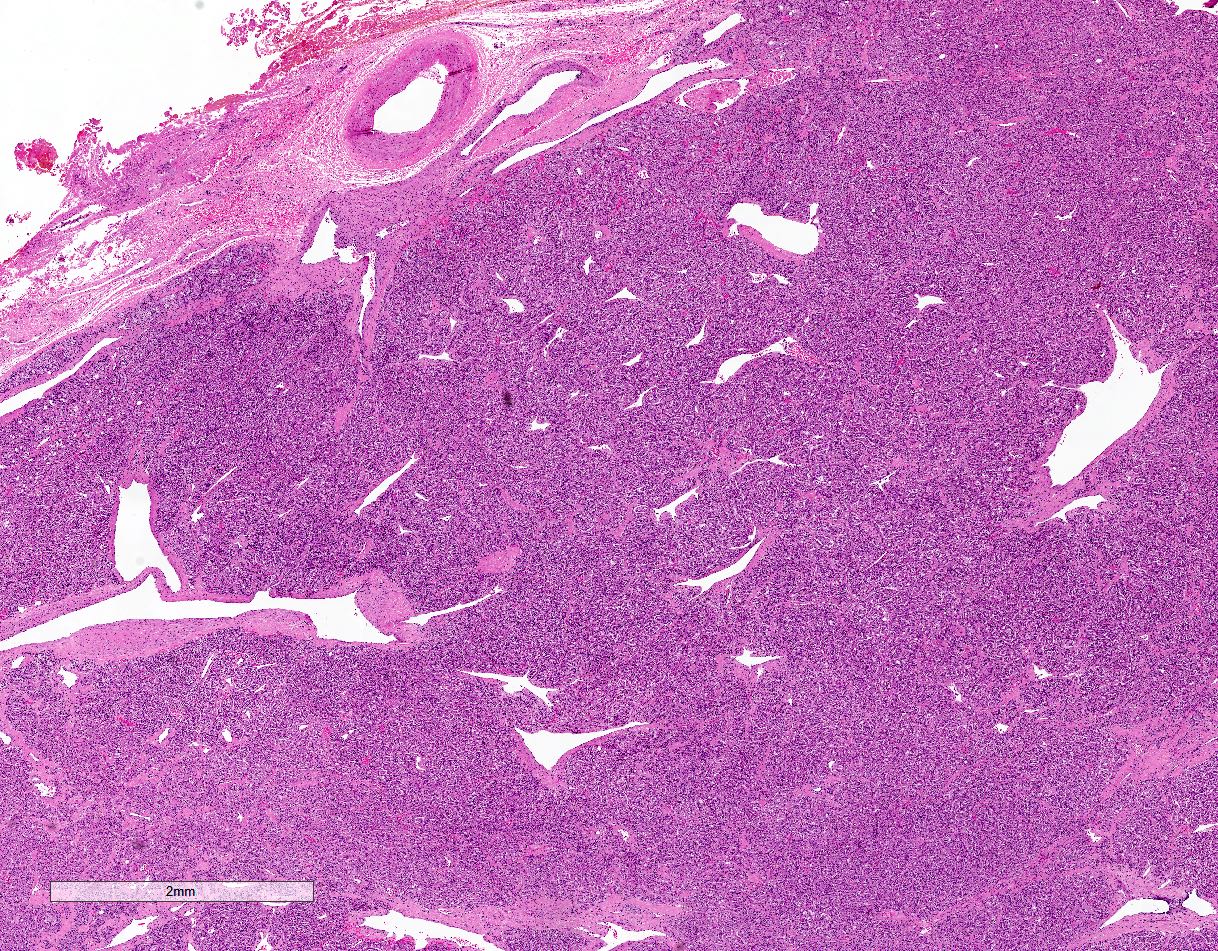
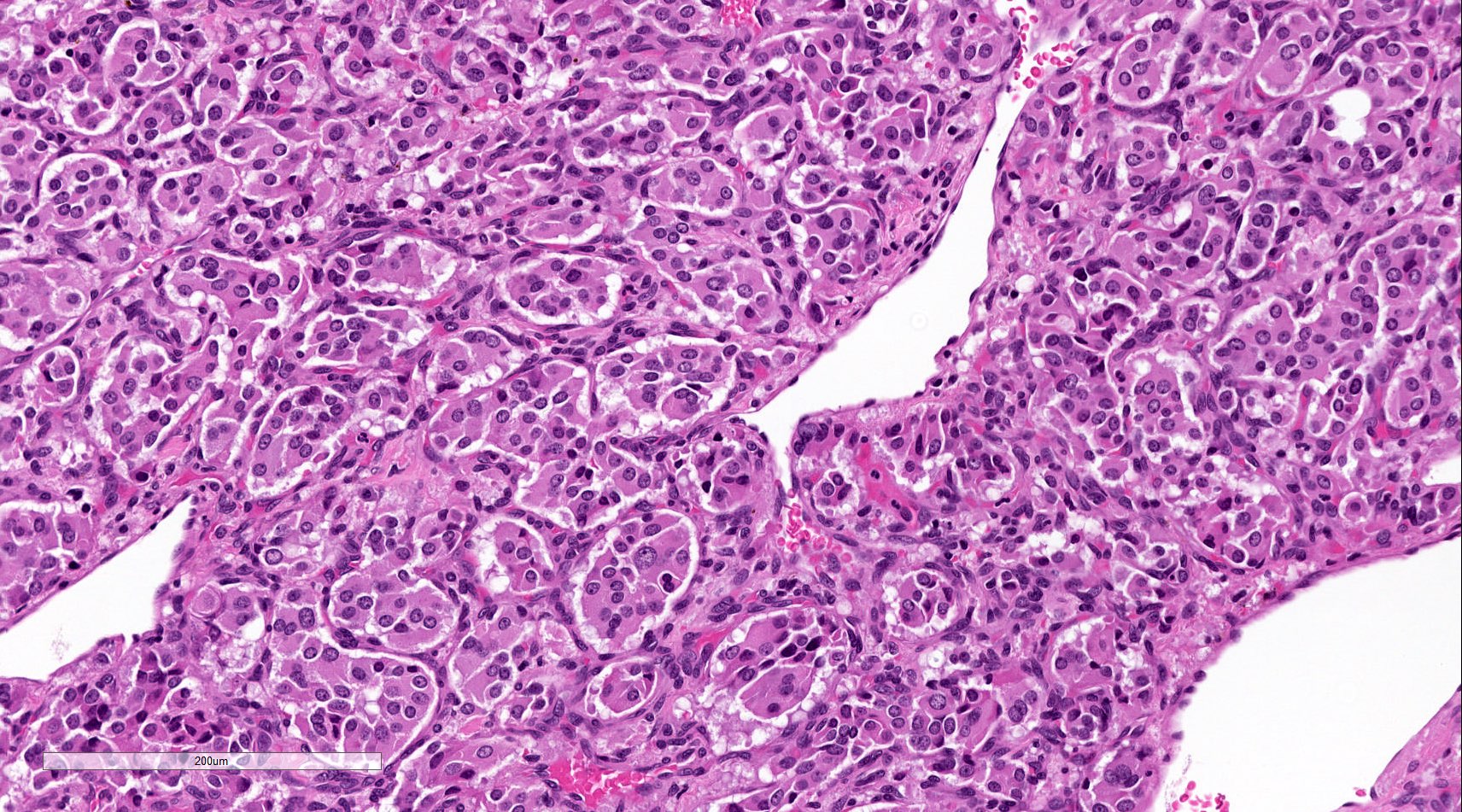
Carotid body paraganglioma
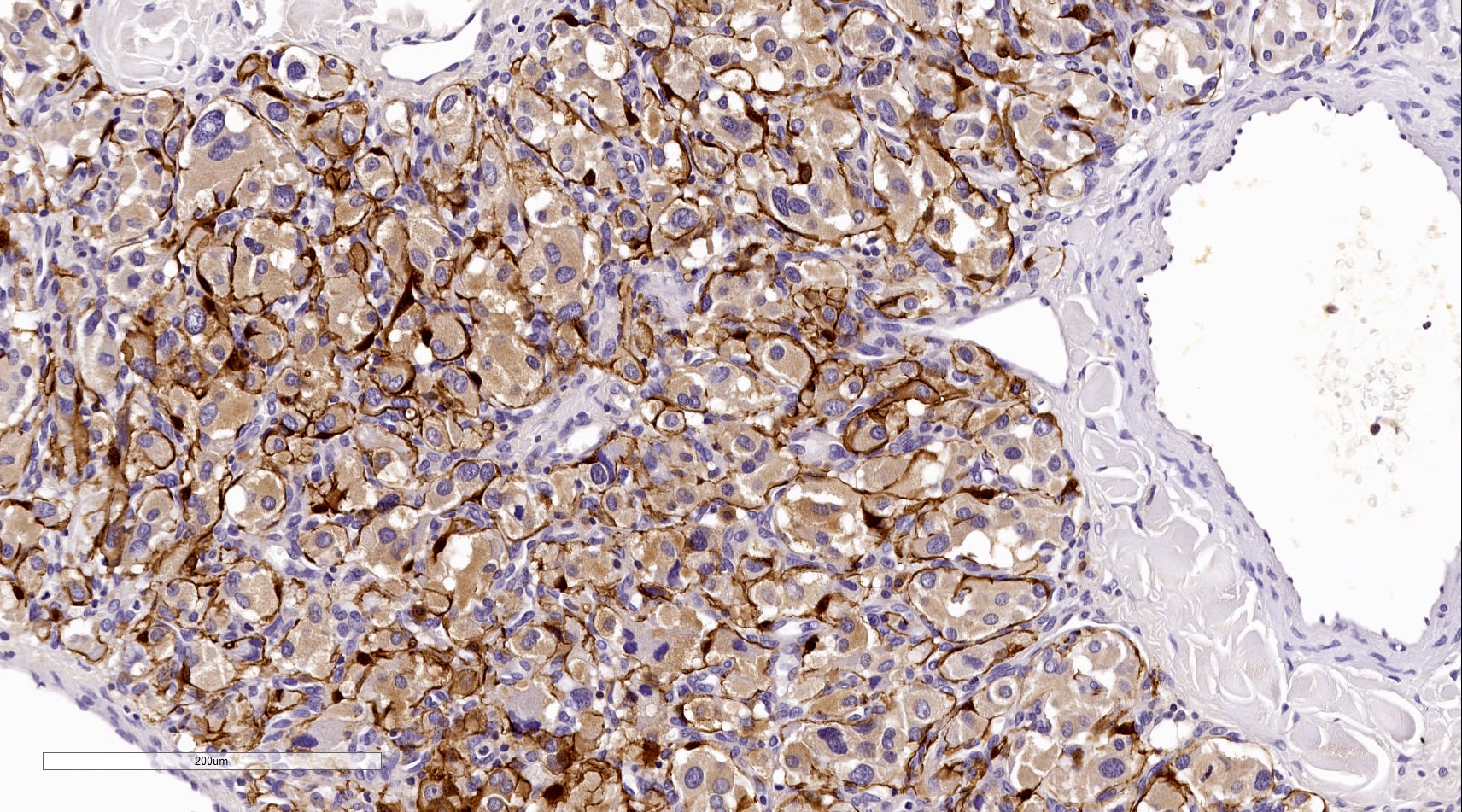
S100
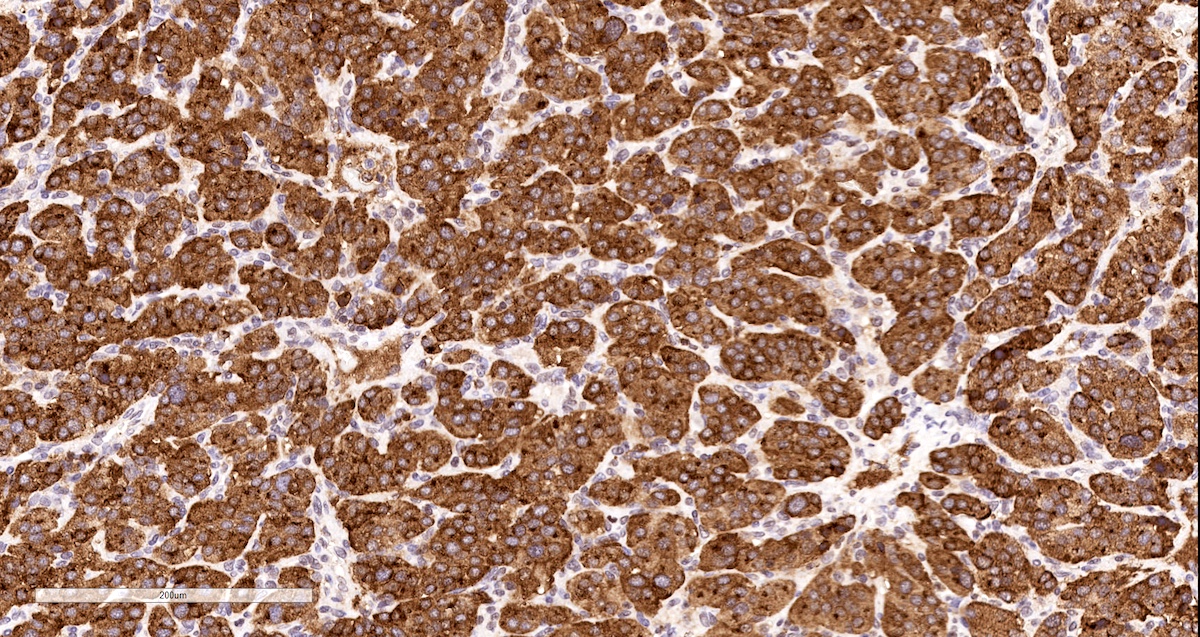
Chromogranin
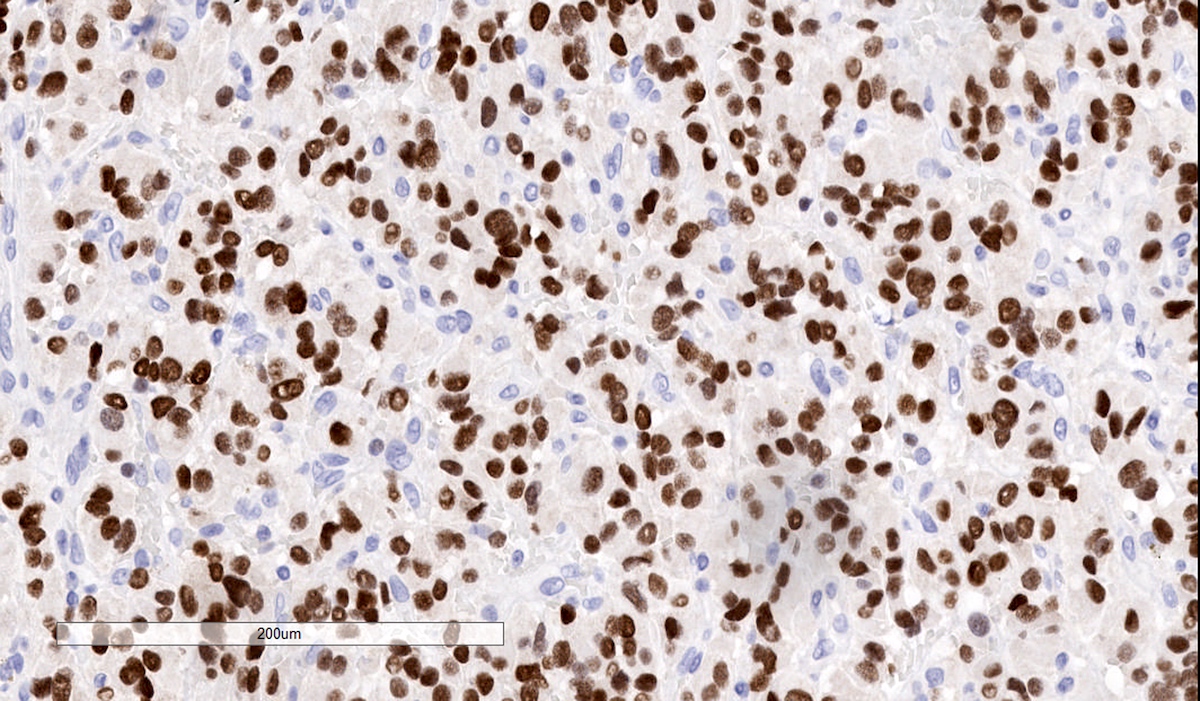
GATA3
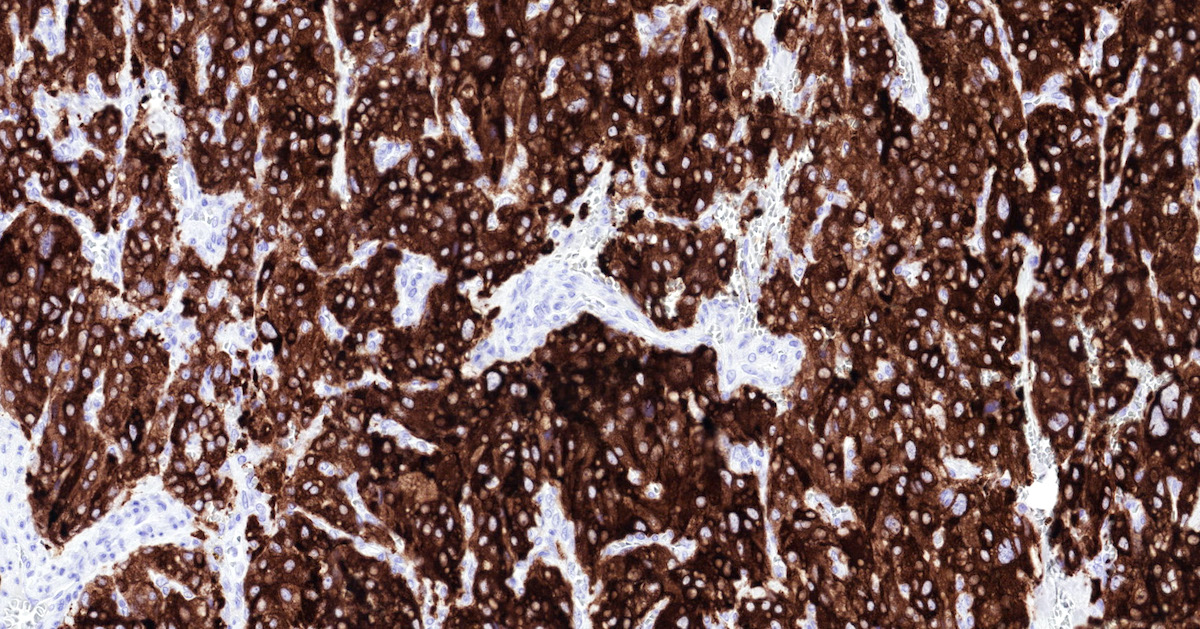
Tyrosine hydroxylase

S100 protein
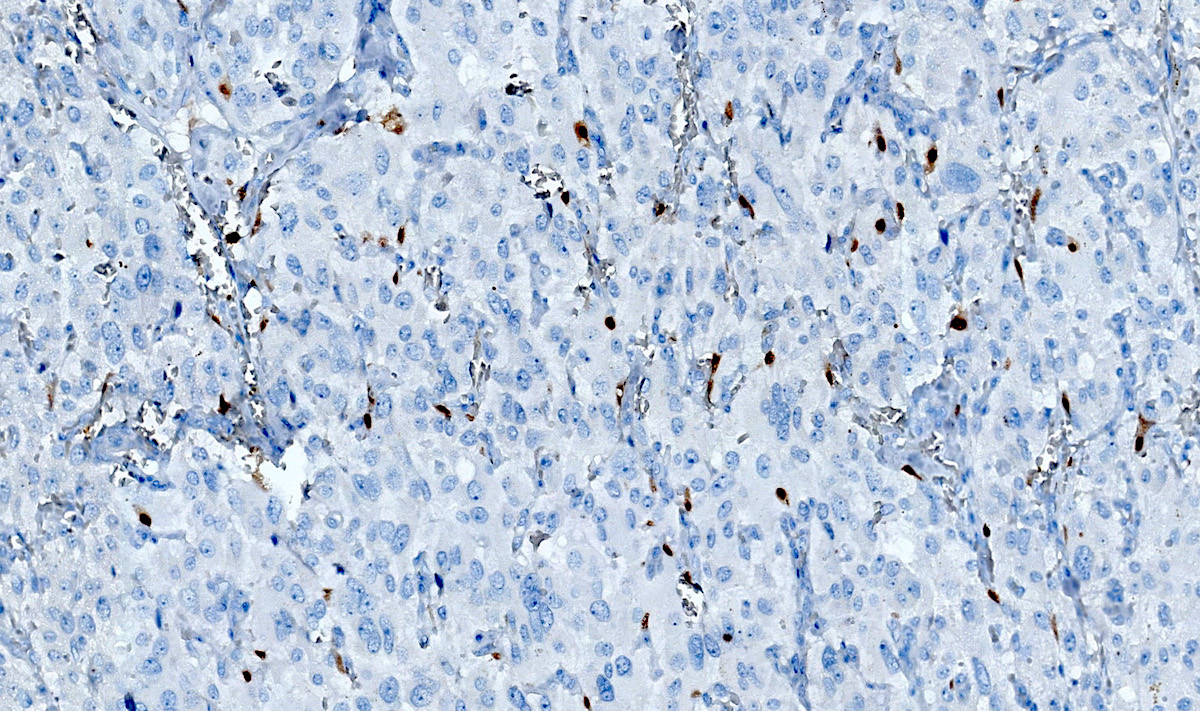
SOX10
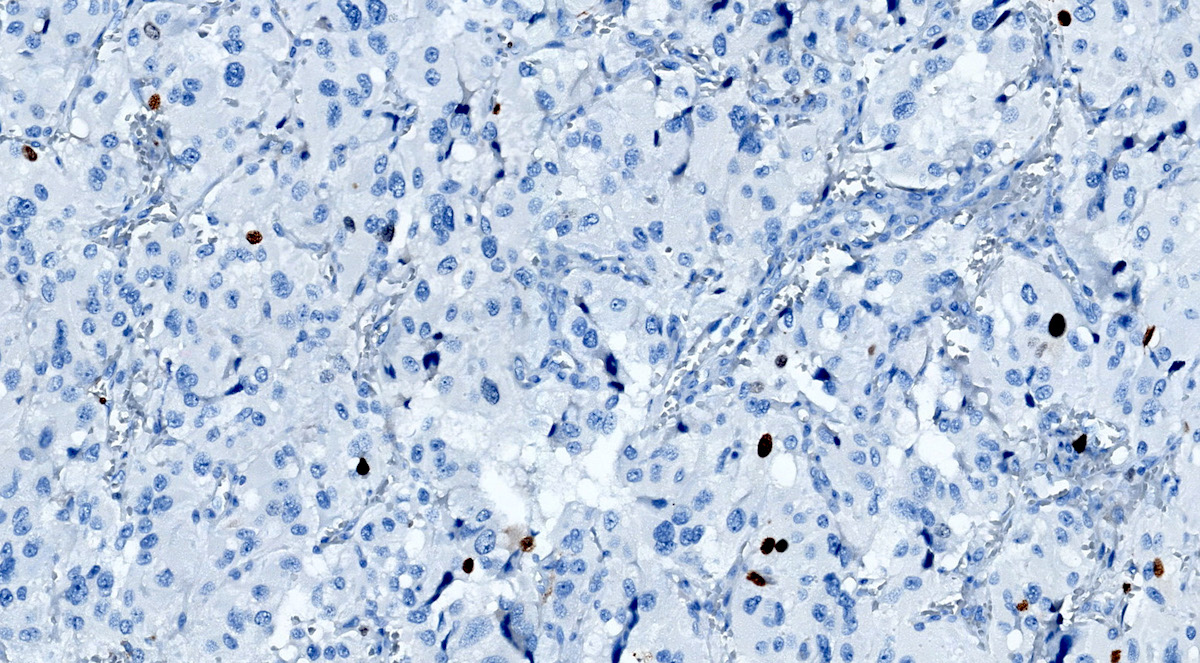
Ki67
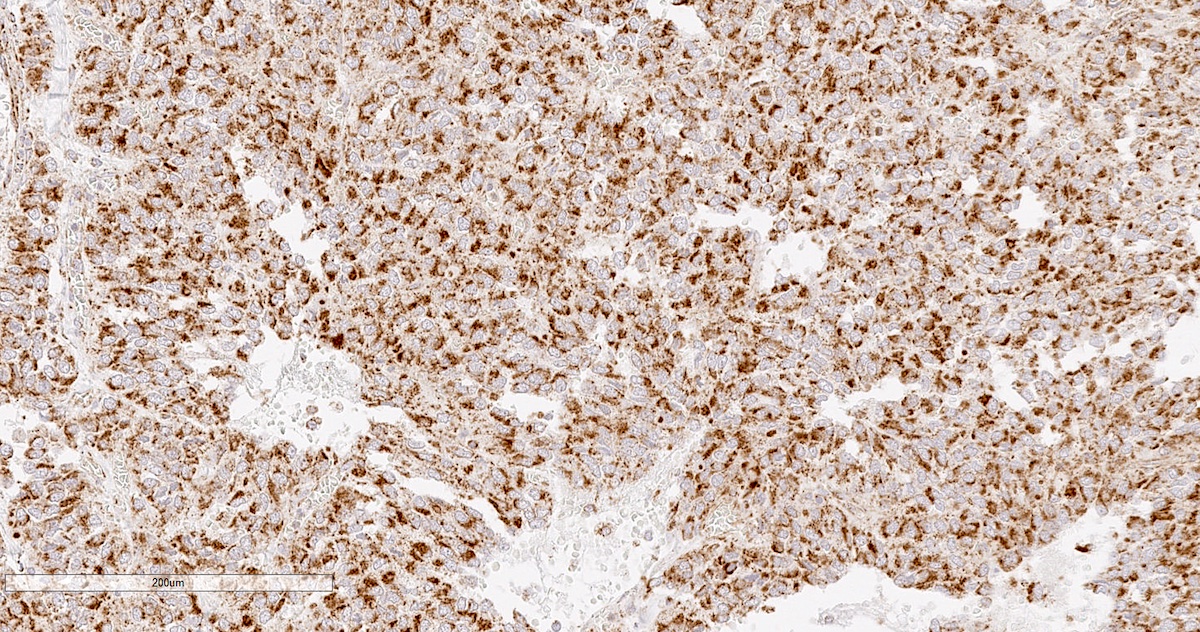
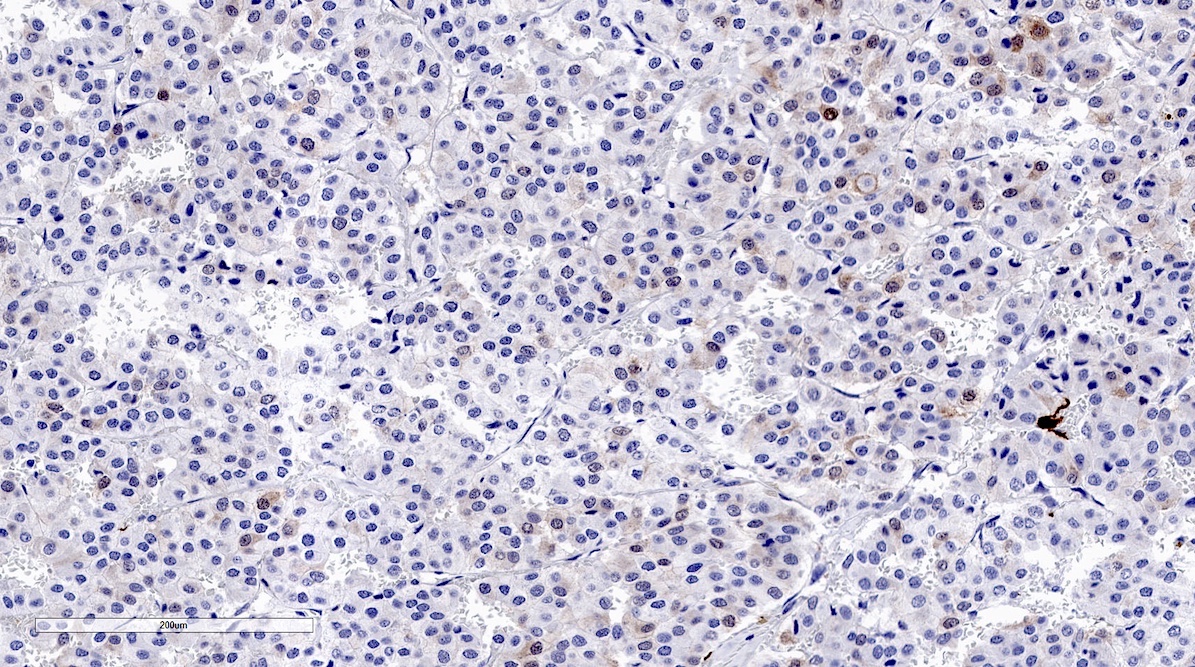
SDHB intact
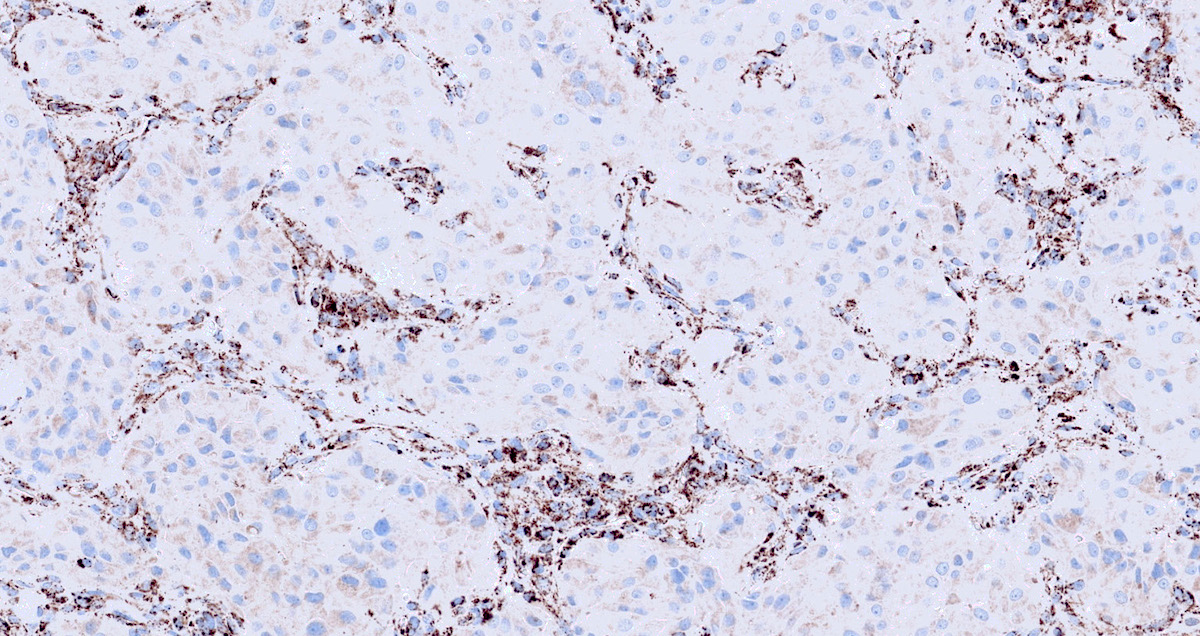
SDHB deficient
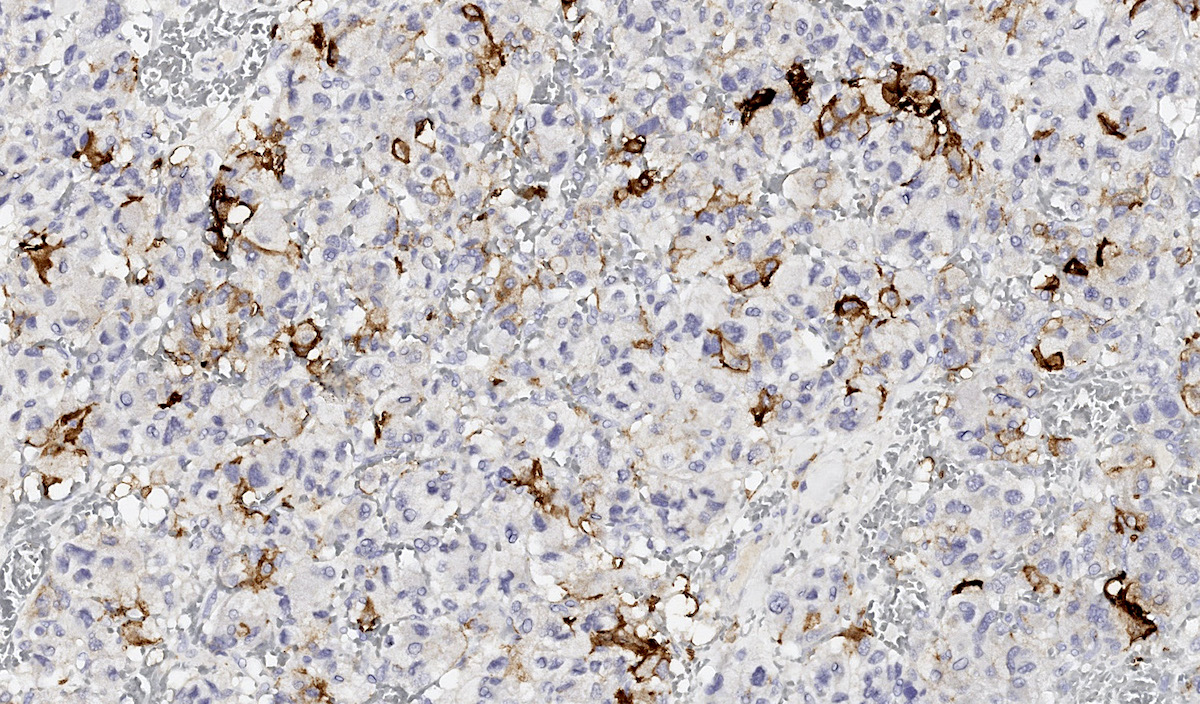
CAIX
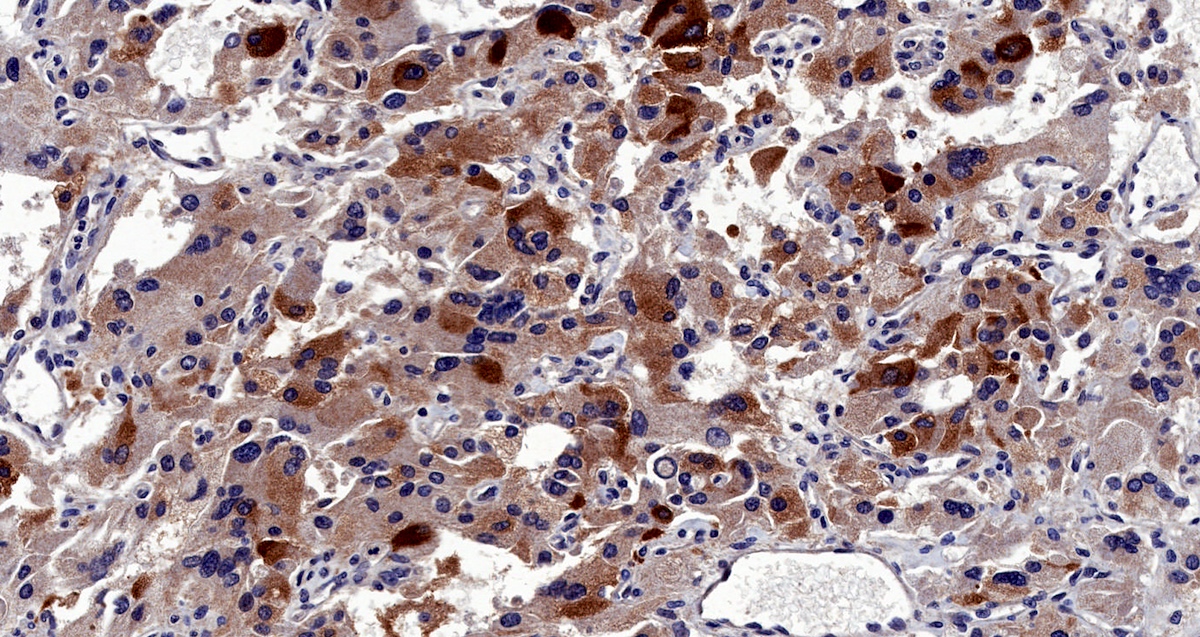
Inhibin
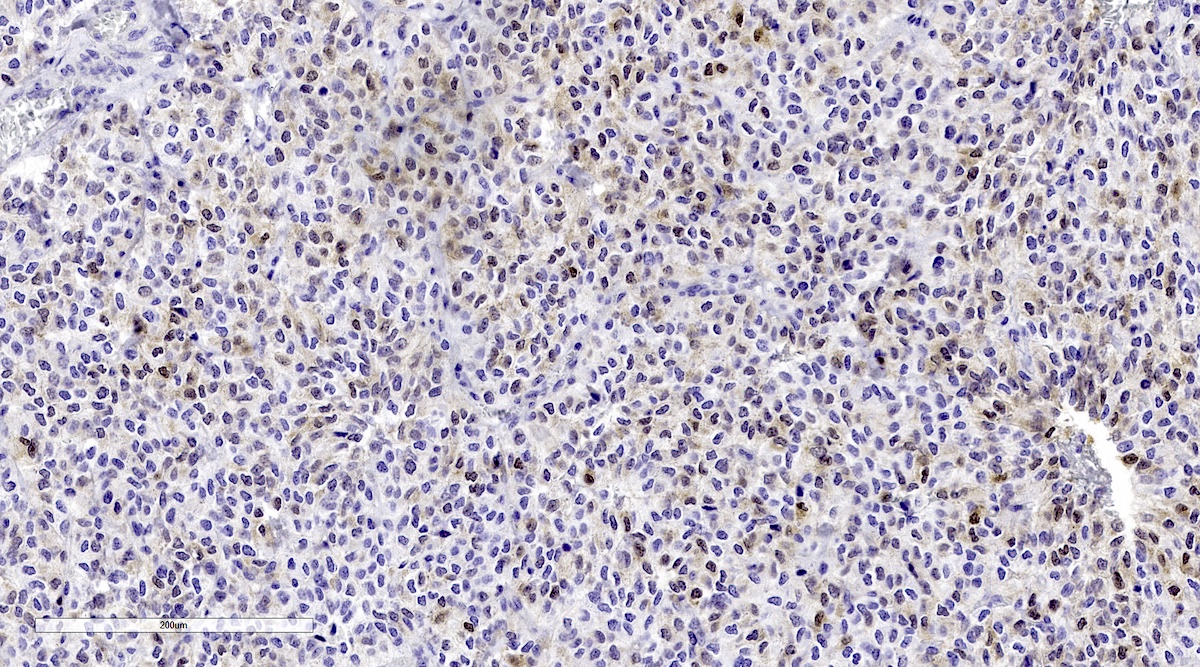
S100 staining in metastatic pheochromocytoma
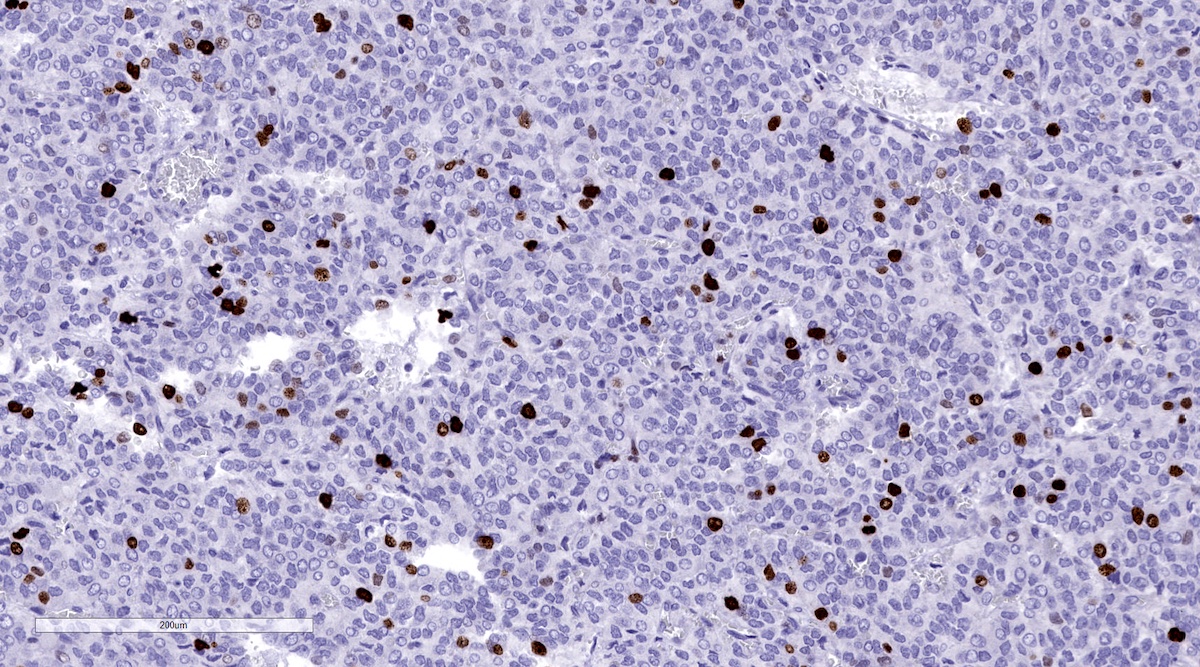
Ki67 in metastatic pheochromocytoma
Cytology description
- Irregular clusters of tumor cells with eosinophilic finely granular cytoplasm and occasional intracytoplasmic pigment (Acta Cytol 2005;49:421)
- Mild to severe nuclear pleomorphism with sporadic binucleation and intranuclear pseudoinclusions (Acta Cytol 2006;50:372)
- Fine needle aspiration not recommended for paragangliomas due to the wide variety of morphologic patterns and high probability of catecholamine crisis and hemorrhage; if required, patients should undergo alpha blocade (Acta Cytol 2003;47:1082)
Positive stains
- INSM1 (diffuse nuclear) (Am J Surg Pathol 2018;42:665)
- Chromogranin A (can be only focal or perinuclear dot-like golgi pattern)
- Synaptophysin (Histol Histopathol 1993;8:429)
- CD56 (Endocr Pathol 2002;13:149)
- S100 may be diffusely positive (J BUON 2018;23:1540)
- GATA3 (diffuse nuclear) (Hum Pathol 2020;103:72, Am J Surg Pathol 2014;38:13, J Clin Med 2018;7:280)
- Tyrosine hydroxylase (critical enzyme required for catecholamine synthesis; usually diffuse and strong in sympathetic paragangliomas, may be weak or focal in parasympathetic tumors) (Hum Pathol 2020;103:72)
- Dopamine beta hydroxylase and phenylethanolamine N methyltransferase (PNMT) (epinephrine producing tumors) (see Diagram below) (Neuroendocrinology 2015;101:289, J Clin Med 2018;7:280)
- Sustentacular cells: S100, GFAP and SOX10 (Hum Pathol 2020;103:72)
- Ectopic immunoexpression of hormones: serotonin, ACTH, CRH, VIP, leu enkephalin substance P, gastrin, somatostatin, vasopressin, MSH and calcitonin (Arch Endocrinol Metab 2017;61:291, J Clin Endocrinol Metab 2021;106:598, Endocr Pract 2014;20:e145, Ann Intern Med 1979;91:208)
- IHC surrogate markers for germline mutations:
- SDHB or SDHA expression loss: germline SDHx mutations (Lancet Oncol 2009;10:764)
- FH expression loss: germline FH mutation (HLRCC) (Hum Pathol 2018;71:47)
- Carbonic anhydrase IX (CAIX) expression: 80% of VHL mutation (Mod Pathol 2020;33:57)
- Inhibin alpha: any hypoxic pathway disease (SDHx, VHL, etc.) (Am J Surg Pathol 2021;45:1264)
- Reticulin highlights the nesting pattern (Int J Gynecol Pathol 1991;10:203, Exp Toxicol Pathol 2013;65:631)
Contributed by Luvy Delfin, M.D. and Sylvia L. Asa, M.D., Ph.D.
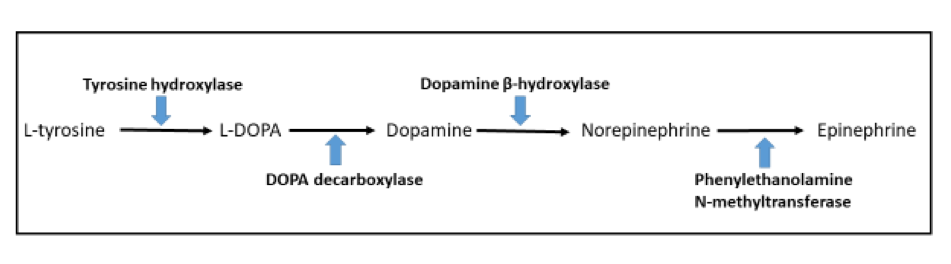
Pathway of catecholamine biosynthesis
Electron microscopy description
- Chief cells are polygonal with well developed rough endoplasmic reticulum, prominent Golgi complex and abundant cytoplasmic neurosecretory granules (Arch Pathol Lab Med 1980;104:46)
- May have giant mitochondria with paracrystalline inclusions
- Sustentacular cells wrap around chief cells and lack neurosecretory granules
- No desmosomes
Electron microscopy images
Contributed by Luvy Delfin, M.D. and Sylvia L. Asa, M.D., Ph.D.
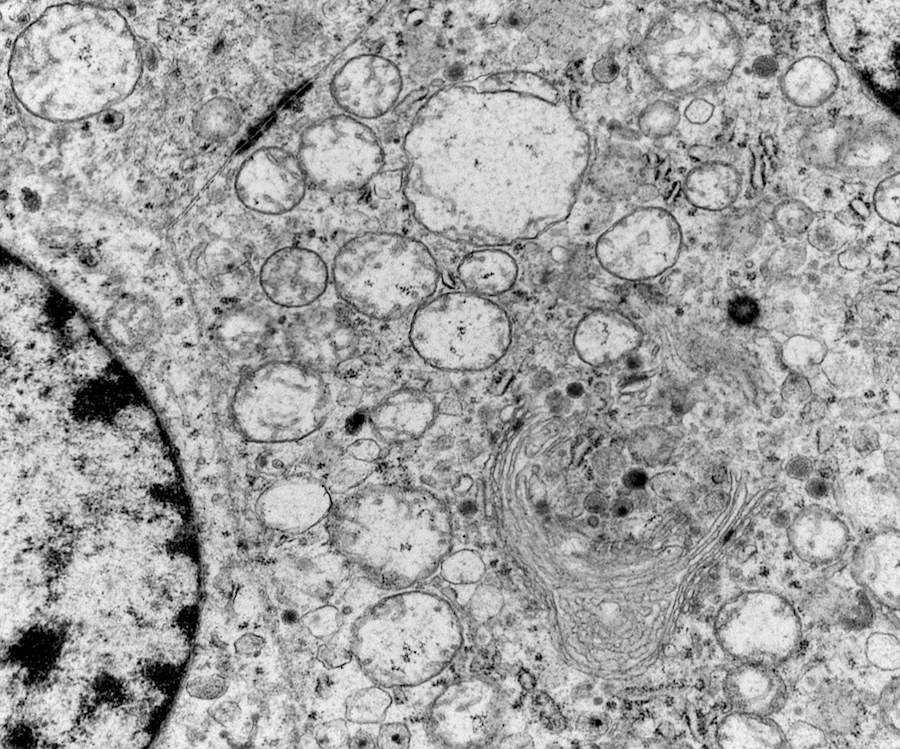
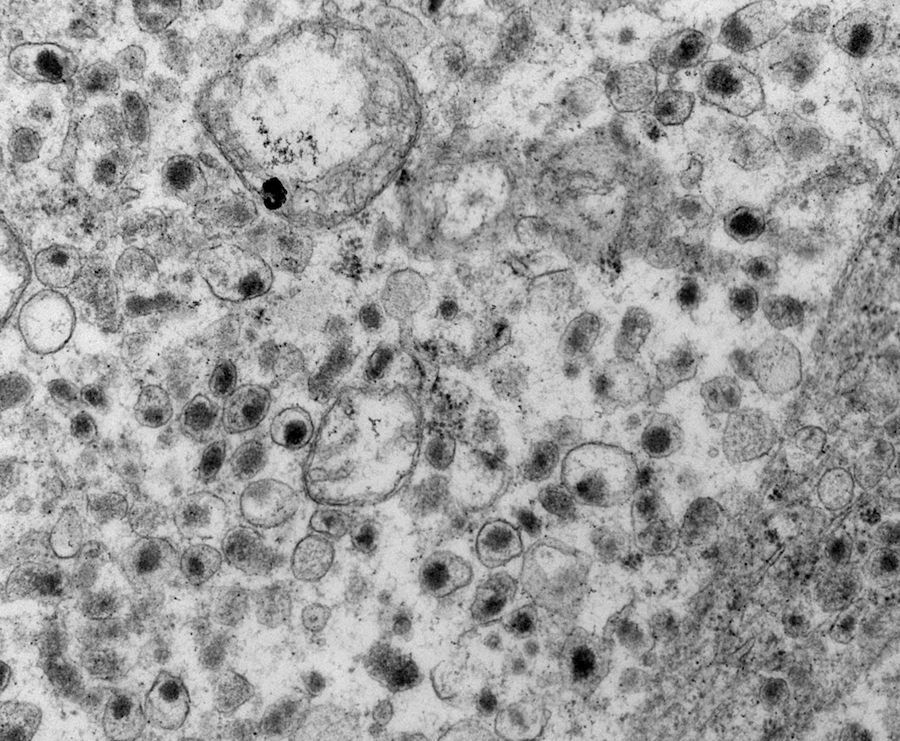
Paraganglioma
Molecular / cytogenetics description
- Transcriptome based classification of paragangliomas (see Table 2) (Cancer Cell 2017;31:181):
- Cluster 1: pseudohypoxia pathway / Krebs cycle related: SDH / FH deficient, mutations in MDH2 and mutant IDH state (1A) and pseudohypoxia pathway / VHL / EPAS1 related (1B): VHL, HIF2A, PHD1 / EGLN2, PHD2 / EGLN1 (dopaminergic or noradrenergic secretory profiles) (Cancers (Basel) 2021;13:3312, Cancer Cell 2017;31:181)
- Cluster 2: kinase signaling (RAS / RAF / ERK, P13 kinase / AKT / mTOR, TMEM127, NF1 and MYC / MAX / MXD1) pathways (adrenergic and noradrenergic secretory profiles)
- Cluster 3: wnt / B catenin pathway activation: CSDE1 or MAML3 somatic mutations (Mol Cancer Res 2021;19:1476, Cancer Cell 2017;31:181)
- Somatic mutations are present in 30% of sporadic paragangliomas: ATRX, TP53, KMT2D, SETD2 and TERT (Int J Endocrinol 2015;2015:138573)
Table 2: molecular clusters of pheochromocytoma and paraganglioma
| Molecular cluster | Mutated gene | Percentage of all PPGLs | % Hereditary |
| Pseudohypoxia TCA cycle related | Succinate dehydrogenase complex iron sulfur subunit (SDHB) | 10 - 15% | 100% |
| Succinate dehydrogenase complex flavoprotein subunit (SDHA) | |||
| Succinate dehydrogenase complex subunit C (SDHC) | |||
| Succinate dehydrogenase complex subunit D (SDHD) | |||
| Succinate dehydrogenase complex assembly factor 2 (SDHAF2) | |||
| Fumarate hydratase (FH) | |||
| Pseudohypoxia, VHL / EPAS1 related | Von Hippel-Lindau tumor suppressor (VHL) | 15 - 20% | 25% |
| Endothelial PAS domain protein 1 (EPAS1) | |||
| Wnt signaling | Cold shock domain containing E1 (CSDE1) | 5 - 10% | 0% |
| Mastermind-like transcriptional coactivator 3 (MAML3) | |||
| Kinase signaling | Ret proto-oncogene (RET) | 50 - 60% | 20% |
| Neurofibromin 1 (NF1) | |||
| MYC associated factor X (MAX) | |||
| Transmembrane protein 127 (TMEM127) | |||
| HRAS proto-oncogene, GTPase (HRAS) |
ICCR guidelines for pheochromocytoma and paraganglioma
- Synoptic reporting should be used (ICCR: Phaeochromocytoma and Paraganglioma [Accessed 20 January 2022])
AJCC staging
- Staging available for pheochromocytoma and sympathetic PPGLs (Amin: AJCC Cancer Staging Manual, 8th Edition, 2017)
Risk stratification (controversial)
- Risk stratification scoring systems proposed when these lesions were classified as benign and malignant:
- Pheochromocytoma of the Adrenal gland Scaled Score (PASS) (Am J Surg Pathol 2002;26:551)
- Grading system for Adrenal Pheochromocytoma and Paraganglioma (GAPP) (Endocr Relat Cancer 2014;21:405)
- Modified GAPP (PLoS One 2017;12:e0187398)
- Composite Pheochromocytoma / Paraganglioma Prognostic Score (COPPS) (Virchows Arch 2019;474:721)
- Validation studies have confirmed that both PASS and GAPP have variable predictive value (J Clin Endocrinol Metab 2020;105:e4661, Cancers (Basel) 2019;11:225)
- Both systems may overestimate risk and have high interobserver variability (Langenbecks Arch Surg 2018;403:785)
- Histological parameters are best interpreted in conjunction with clinical and molecular data (Hum Pathol 2021;110:8)
- Prognosis depends on complete tumor resection
- Considered histological parameters: histologic pattern, cellularity, comedo type necrosis and capsular / vascular invasion
- Immunohistochemical assessment: Ki67 labeling index
- Biochemical data: catecholamine type
- Clinical characteristics of potentially aggressive pheochromocytoma and paraganglioma, North American Neuroendocrine Tumor Society guidelines 2021 (Pancreas 2021;50:469)
- Presurgical:
- Tumor size > 5 cm if located in adrenal medulla and > 4 cm if extra-adrenal
- Gross vessel invasion
- Germline SDHB pathogenic variant
- Postsurgical:
- Tumor with adjacent lymph node involvement
- Persistently elevated metanephrine levels
- High mitotic index or Ki67 labeling index
- Presurgical:
Sample pathology report
- Adrenal (right), adrenalectomy:
- Pheochromocytoma, 6 cm (see synoptic report)
- Synoptic report: pheochromocytoma and extra-adrenal paraganglioma
- Procedure: adrenalectomy - right
- Biochemical features: biochemically functioning
- Metanephrine or adrenaline
- Normetanephrine or noradrenaline
- Dopamine
- Tumor location and size (by imaging)
- Anatomic location: right adrenal
- Greatest dimension: 4 cm
- Additional dimensions: 3.8 cm
- Specimen description
- Received: in formalin
- Specimen integrity: intact
- Specimen size : 6 x 6 x 3.5 cm
- Specimen weight: 56.2 grams
- Tumor focality: unifocal
- Tumor size: 6 x 4 x 3.5 cm
- Tumor type: pheochromocytoma
- Histologic features:
- Growth pattern: nested (alveolar, zellballen) pattern
- Composite tumor elements: absent
- Cytologic variants: epithelioid
- Necrosis: not identified
- Mitotic rate: < 2 mitoses (per 10 high power fields or mm2)
- Additional features: none
- Encapsulation: no capsule
- Invasive growth
- Tumor capsule invasion: not applicable
- Adrenal capsule invasion: not identified
- Surrounding tissues: not identified
- Vascular invasion: not identified
- Lymphatic invasion: not identified
- Surgical margins: uninvolved
- Metastases
- Lymph nodes: not identified
- Distant: not assessed
- Immunohistochemistry
- Chromogranin A: positive
- GATA3: positive
- Tyrosine hydroxylase: positive
- S100 protein: positive in tumor cells and strong in sustentacular cells
- SOX10: positive in sustentacular cells
- SDHB: intact
- CAIX: negative
- Inhibin: negative
- Ki67 10%
- Associated lesions
- Adrenal medullary hyperplasia: none
- Current or past tumors in other organs (specify): none
- Gross description example
- Received in formalin, labeled with the patient's name and hospital number and right adrenal gland, are multiple fragments of fibroadipose tissue with an adrenal gland containing a mass; the fragments measure 6.0 x 6.0 x 3.5 in aggregate and weigh 56.2 g. The surface is inked black. The adherent fat is removed and the adrenal gland with mass weighs 43.5 g. The mass measures 6.0 x 4.0 x 3.5 cm and weighs 36.6 g. The cut surface of the mass reveals a well delineated soft homogenous surface with central hemorrhage. The nontumorous adrenal measures 3.0 x 1.5 x 1.0 cm and has an unremarkable cut surface. In the periadrenal fibroadipose tissue there are no nodules or lymph nodes. A photograph is taken. Representative sections are submitted in 26 cassettes.
- 1 - 3 mass with portion of adrenal gland, serially sectioned
- 4 - 8 complete cross section of mass
- 9 - 20 capsule entirely submitted
- 21 - 23 mass with portion of adrenal gland, serially sectioned
- 24 - 26 nontumorous adrenal entirely submitted
Differential diagnosis
- Head and neck region:
- Neuroendocrine tumor / neuroendocrine carcinoma:
- Generally positive for keratins and specific site origin transcription factors, negative on PGLs (J Clin Med 2018;7:280)
- Medullary thyroid carcinoma (Curr Oncol 2019;26:338):
- Usually solid nests of C cells within a vascular stroma, variable amyloid deposition
- CK+, CEA+, calcitonin+, variable TTF1, synaptophysin+, chromogranin+
- GATA3-, tyrosine hydroxylase- with rare exceptions (Endocr Pathol 2017;28:362)
- Parathyroid adenoma (Endocr Pathol 2018;29:113):
- May have nested pattern also solid, follicular, acinar, cords, rosette-like
- Composed by chief, oxyphilic transitional or clear cells
- CK+, CAM5.2+, GATA3+, PTH+, chromogranin A+ and intact parafibromin
- S100-, tyrosine hydroxylase-
- Middle ear neuroendocrine tumor / middle ear adenoma (Endocr Pathol 2021 May 27 [Epub ahead of print]):
- Mainly interlacing ribbons and tubules of low cuboidal or columnar cells
- CK+, INSM1+, synaptophysin+, chromogranin+, human pancreatic polypeptide+, glucagon+, SATB2+, PSAP variable
- S100-, SOX10-, tyrosine hydroxylase-
- Pituitary neuroendocrine tumor (PitNET) (Mod Pathol 2019;32:484):
- Meningioma, ear and temporal bone (primary intra-tympanic / temporal bone / cerebellopontine angle / skull base) (J Laryngol Otol 2006;120:786, Lin Chung Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 2016;30:662, Head Neck 2007;29:793):
- Schwannoma, extracranial head and neck locations (J Cancer Res Ther 2019;15:659):
- Ependymoma, skull base, ectopic or craniospinal dissemination (Pathol Int 2005;55:781, Int J Neurosci 2021;131:919):
- Gliofibrillar background, monomorphic cells, round to oval nuclei
- True rosettes and perivascular pseudorosettes
- Vimentin+, GFAP+, S100+ (diffuse), EMA+ (perinuclear dot-like), CD99+
- Synaptophysin-, chromogranin A-
- Neuroendocrine tumor / neuroendocrine carcinoma:
- Thoracic and abdominal region:
- Neuroendocrine tumor / neuroendocrine carcinoma (Surg Pathol Clin 2020;13:35):
- Microacinar, trabecular or ribbon-like growth patterns
- Positive for pankeratins and neuroendocrine markers
- Positive for specific site origen transcription factors, which are negative on PGLs
- Negative for tyrosine hydroxylase
- Ectopic parathyroid tumor (Int J Surg Case Rep 2019;58:153, Front Endocrinol (Lausanne) 2020;11:647):
- Parathyroid adenoma as described above
- Parathyroid carcinoma; infiltrative growth pattern, increase mitotic rate, extensive fibrosis, capsular, vascular or perineural invasion absent on PGLs
- Positive for pankeratin and PTH
- Also positive for neuroendocrine markers and GATA3
- Alveolar soft part sarcoma (Rare Tumors 2018;10:2036361318810907):
- Most common on soft tissue of lower extremities, rarely trunk, internal organs and head and neck (orbit and tongue, usually children)
- Negative for neuroendocrine markers and positive for cathepsin K and TFE3
- Intracytoplasmic rod-like / rhomboid PAS positive / diastase resistant crystalline structures
- ASPSCR1-TFE3 translocation: nuclear immunoreactivity for TFE3
- Gastrointestinal stromal tumor (GIST) (Pathologica 2021;113:230):
- Spindled, epitheliod or mixed type histologically
- Loss of SDHB protein expression (SDH deficient GIST) (Arch Pathol Lab Med 2020;144:655)
- CD117+, DOG1+, CD34+ (75%), h-caldesmon+
- KIT or PDGFRA mutations in up to 90% of cases
- Granular cell tumor (Ann Gastroenterol 2018;31:439):
- Nonencapsulated submucosal sheets, nests and cords of polygonal cells with abundant eosinophilic granular cytoplasm, no characteristic zelballen pattern
- Strong diffuse positive S100, SOX10 contrary to scattered sustentacular cell of PGLs
- Also calretinin+, CD68+, inhibin A+ and PAS+ cytoplasmic granules
- Negative for neuroendocrine markers and keratin
- Malignant gastrointestinal neuroectodermal tumor (Am J Surg Pathol 2020;44:456):
- Rare, most common in small intestine submucosa
- Sheet-like or nested proliferation of epithelioid or spindle cells with scattered osteoclast type multinucleated giant cells (Am J Surg Pathol 2012;36:857)
- Diffuse strong S100+ and SOX10+, contrary to scattered sustentacular cell of PGLs
- Occasional synaptophysin+, CD56+, NSE+ but negative for tyrosine hydroxylase
- EWSR1 gene rearrangements with CREB1 or ATF1
- Alveolar rhabdomyosarcoma:
- Most common in deep soft tissues of extremities: also head and neck, trunk, paraspinal, perineal, retroperitoneum
- Some morphology overlap (nested, pseudoalveolar growth pattern) but with high mitotic index and multinucleated tumor giant cells
- Usually diffuse desmin+, myogenin+, MyoD1+
- Variable expression of CD56, synaptophysin, S100 (not sustentacular cell-like) and CK (Mod Pathol 2008;21:795, Head Neck Pathol 2018;12:181)
- Characteristic translocations: t(2;13)(PAX3-FOXO1) or t(1;13)(PAX7-FOXO1)
- Neuroendocrine tumor / neuroendocrine carcinoma (Surg Pathol Clin 2020;13:35):
- Melanoma (J Cutan Pathol 2008;35:433):
- Common epitheliod nested or spindled pattern
- Marked nuclear pleomorphism and prominent nucleoli and mitotic rate
- Generally strong and diffuse S100, contrary to scattered sustentacular cell of PGLs
- Melanocytic markers: MelanA+, HMB45+, SOX10+, PRAME+, MITF+, cathepsin K+
- Rarely positive for neuroendocrine markers (Ultrastruct Pathol 2020;44:249, J Med Case Rep 2020;14:44)
- Adrenal:
- Adrenal cortical adenoma and adrenal cortical carcinoma (Surg Pathol Clin 2019;12:967):
- Sheets or small nests and cords of vacuolated clear cells or oncocytic cells
- Adrenal carcinoma: capsular and lymphovascular invasion, necrosis, mitosis
- Synaptophysin+ (not useful on the differential diagnosis)
- SF1+, MelanA+, calretinin+, inhibin A+ (but also in hypoxic PPGLs (Am J Surg Pathol 2021;45:1264)
- Negative: tyrosine hydroxylase and chromogranin A, GATA3
- Metastatic carcinomas:
- CK+, other tissue specific markers depending on origin, negatives on PGLs
- Most common origin: lung, kidney, breast, melanoma
- Adrenal cortical adenoma and adrenal cortical carcinoma (Surg Pathol Clin 2019;12:967):
Practice question #1
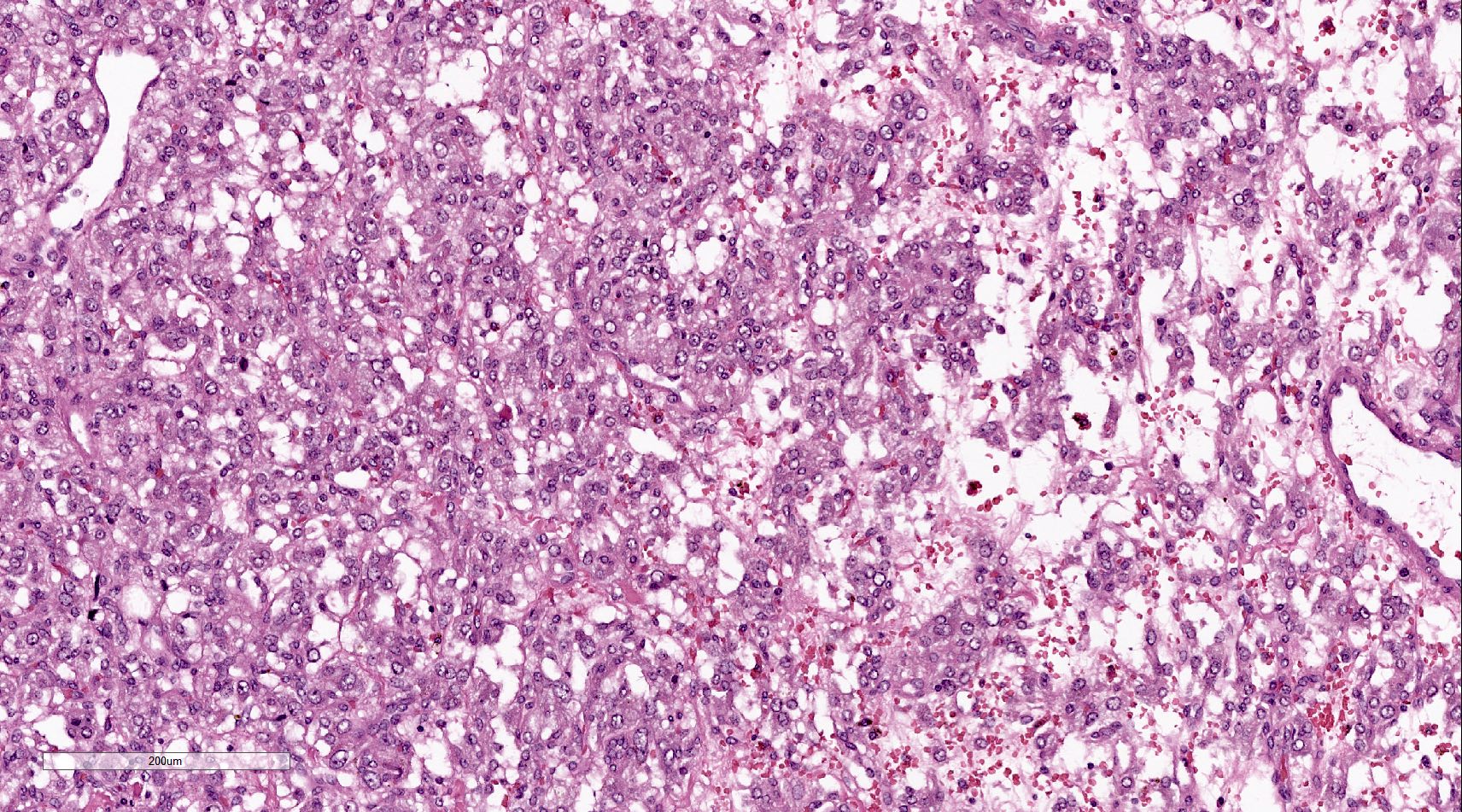
The presence of which of the following histologic features in pheochromocytomas can be associated with von Hippel-Lindau syndrome?
- Bizarre giant cells
- Hyaline droplets / globules
- Prominent stromal edema, clear cystoplasm and lipid degeneration
- Stromal amyloid
Practice answer #1
C. Prominent stromal edema, clear cytoplasm and lipid degeneration in the tumor are usually asscociated with von Hippel-Lindau syndrome
Comment Here
Reference: Paraganglioma
Comment Here
Reference: Paraganglioma
Practice question #2
The strongest genetic risk factor for the development of metastatic paraganglioma is
- NF1
- RET oncogene
- SDHB mutations
- THEM127 and VHL
Practice answer #2
C. Presence of constitutional SDHB mutation is the strongest genetic risk factor for the development of metastasis
Comment Here
Reference: Paraganglioma
Comment Here
Reference: Paraganglioma
