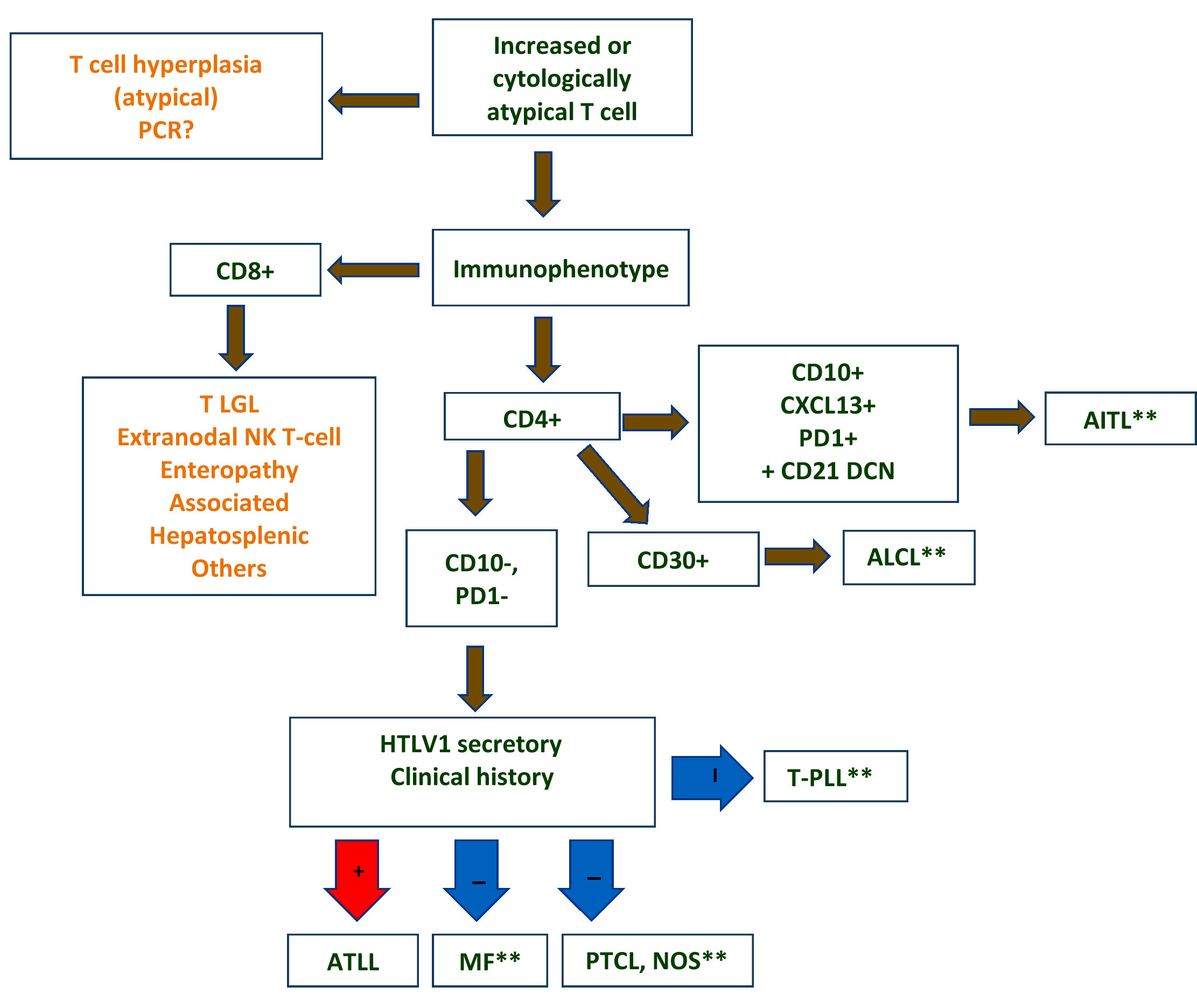
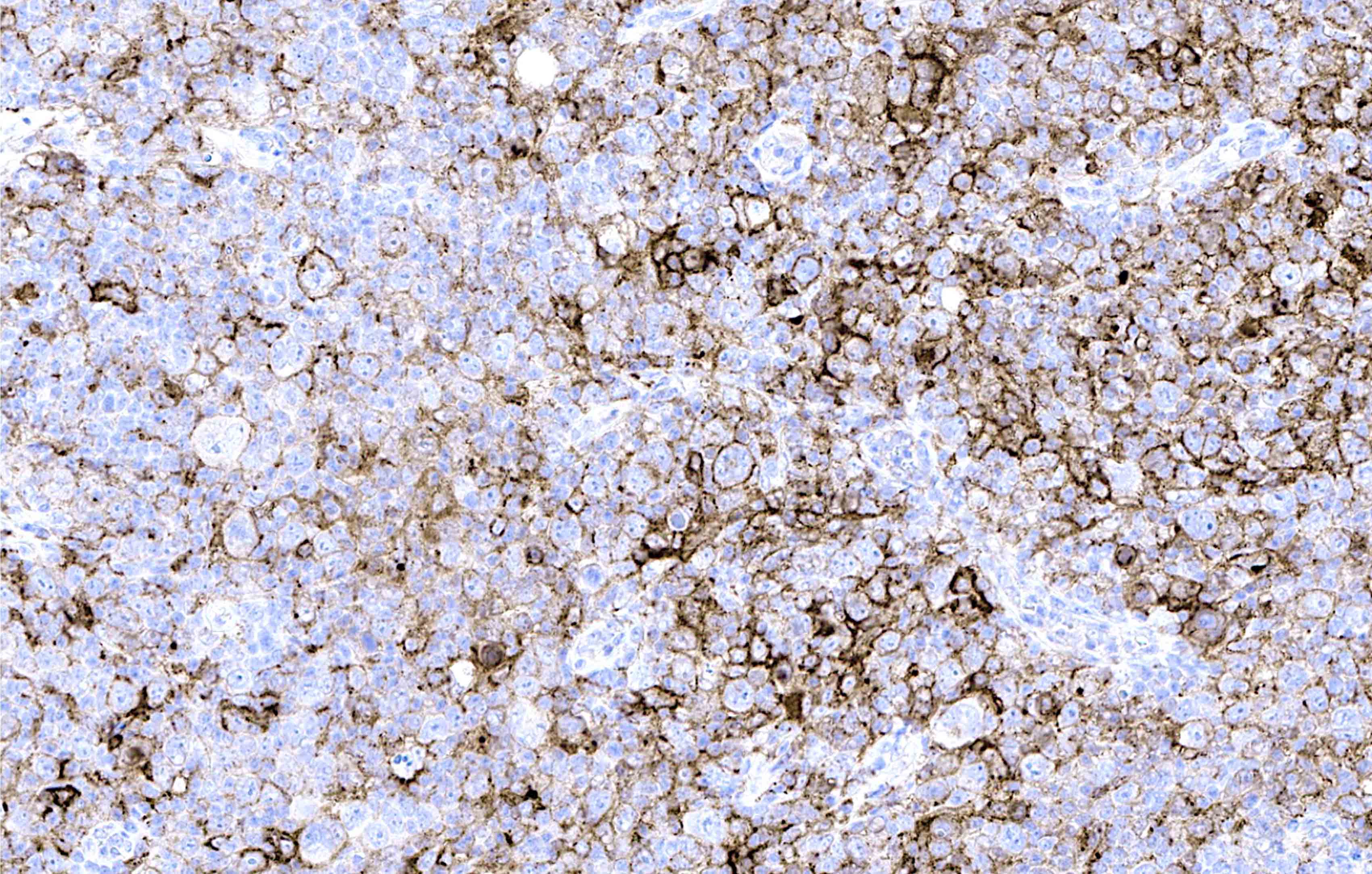
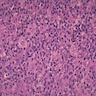
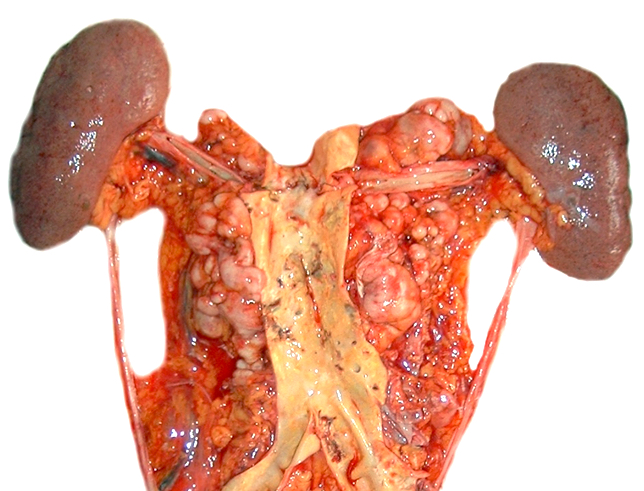
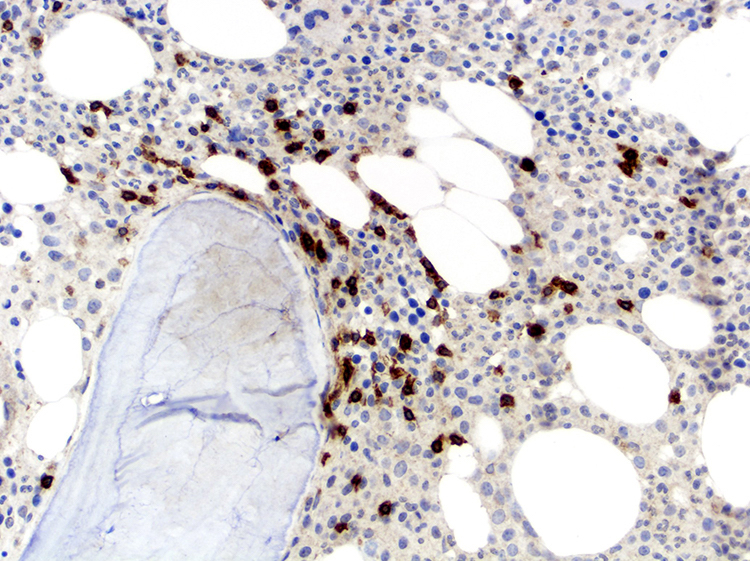
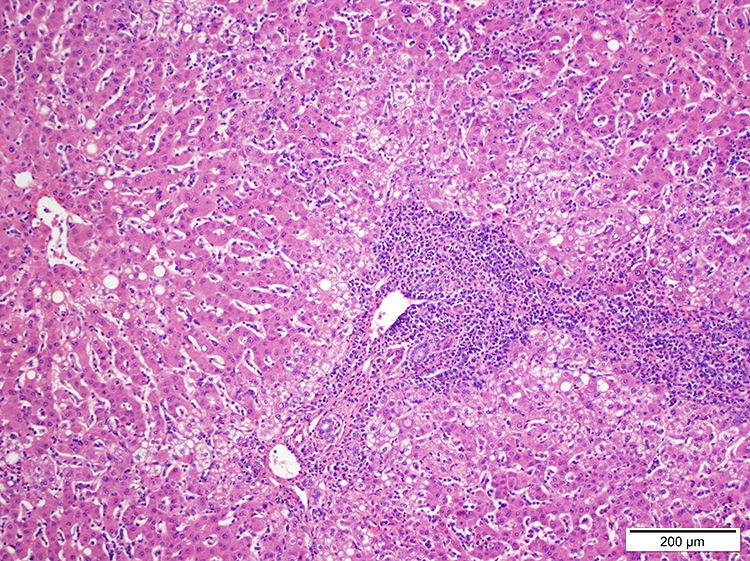
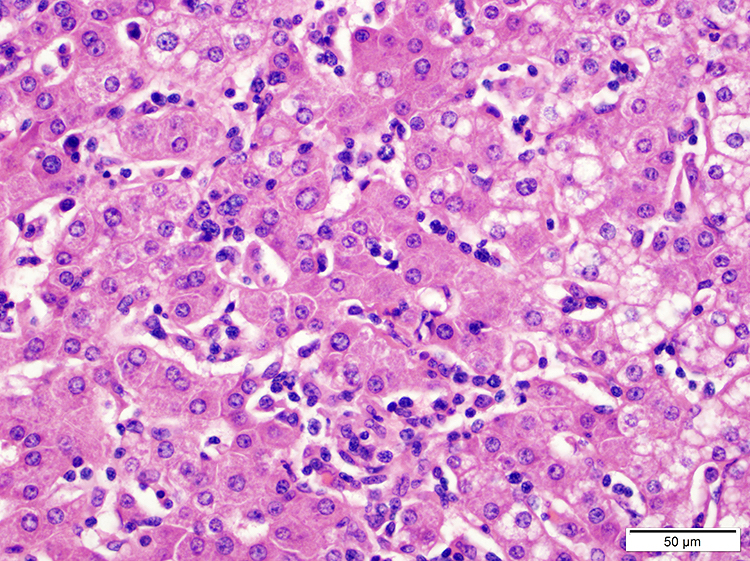
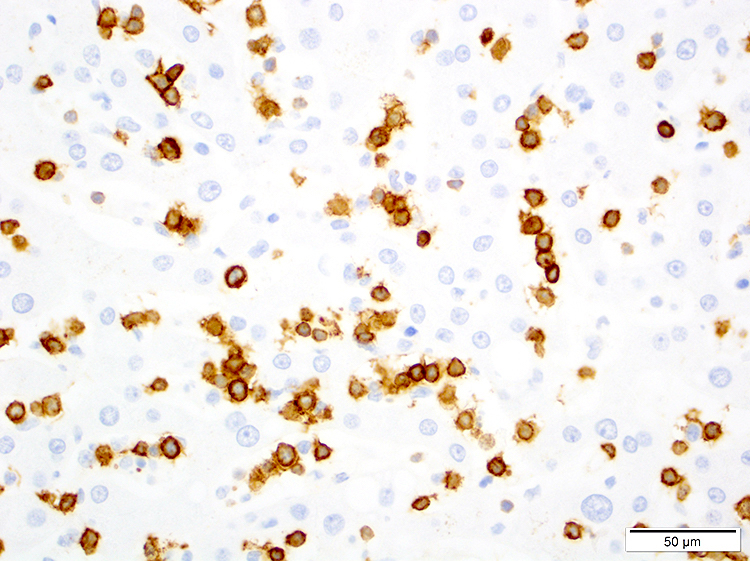
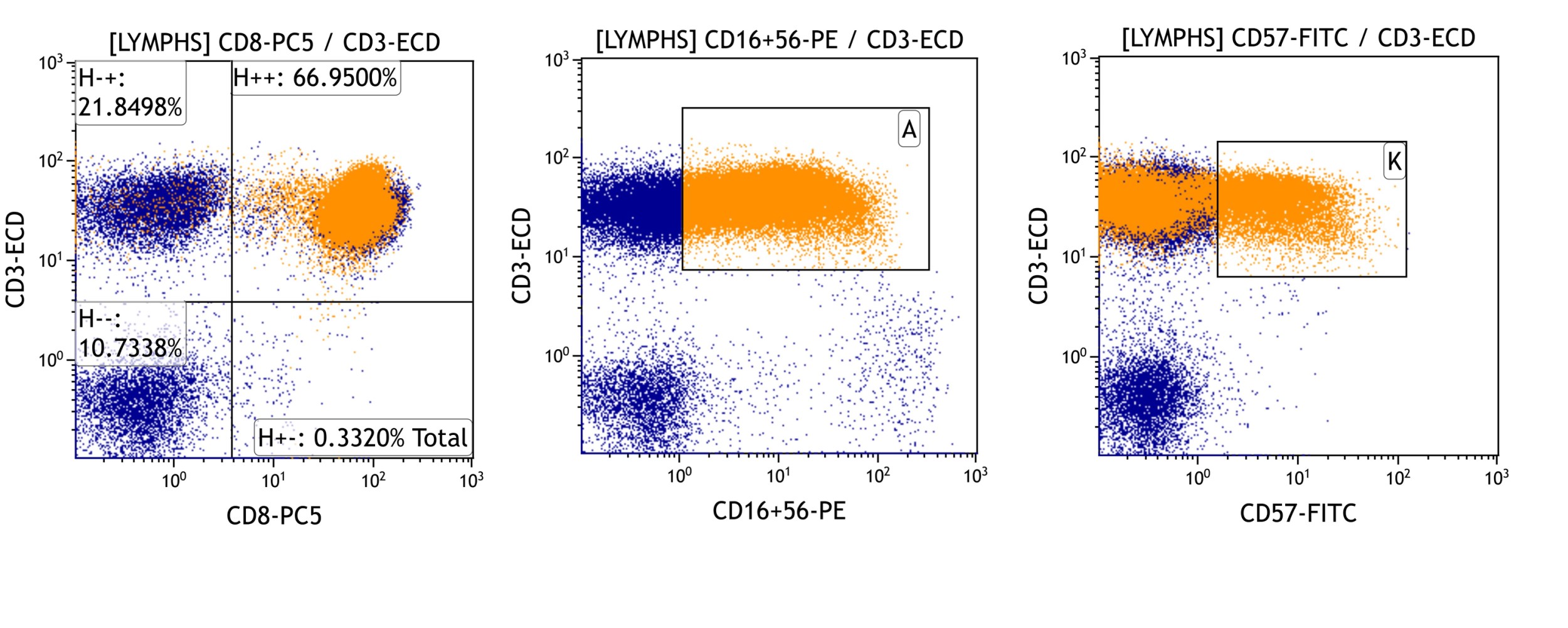
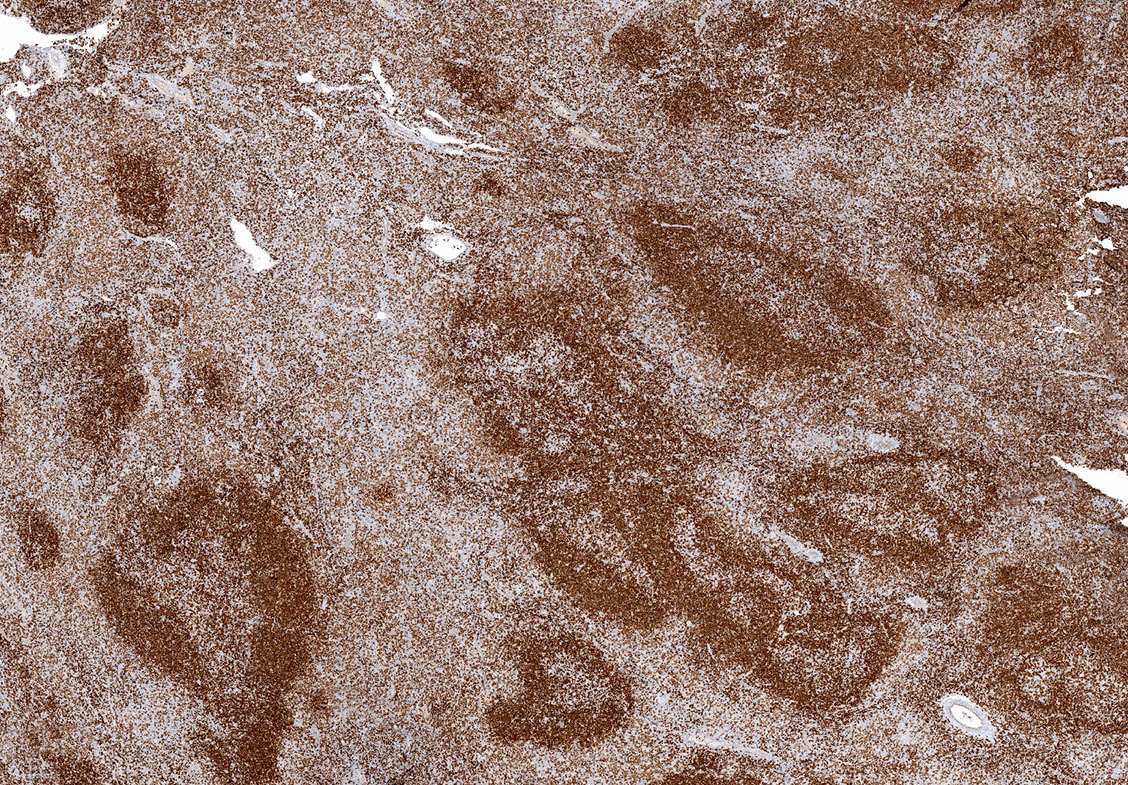
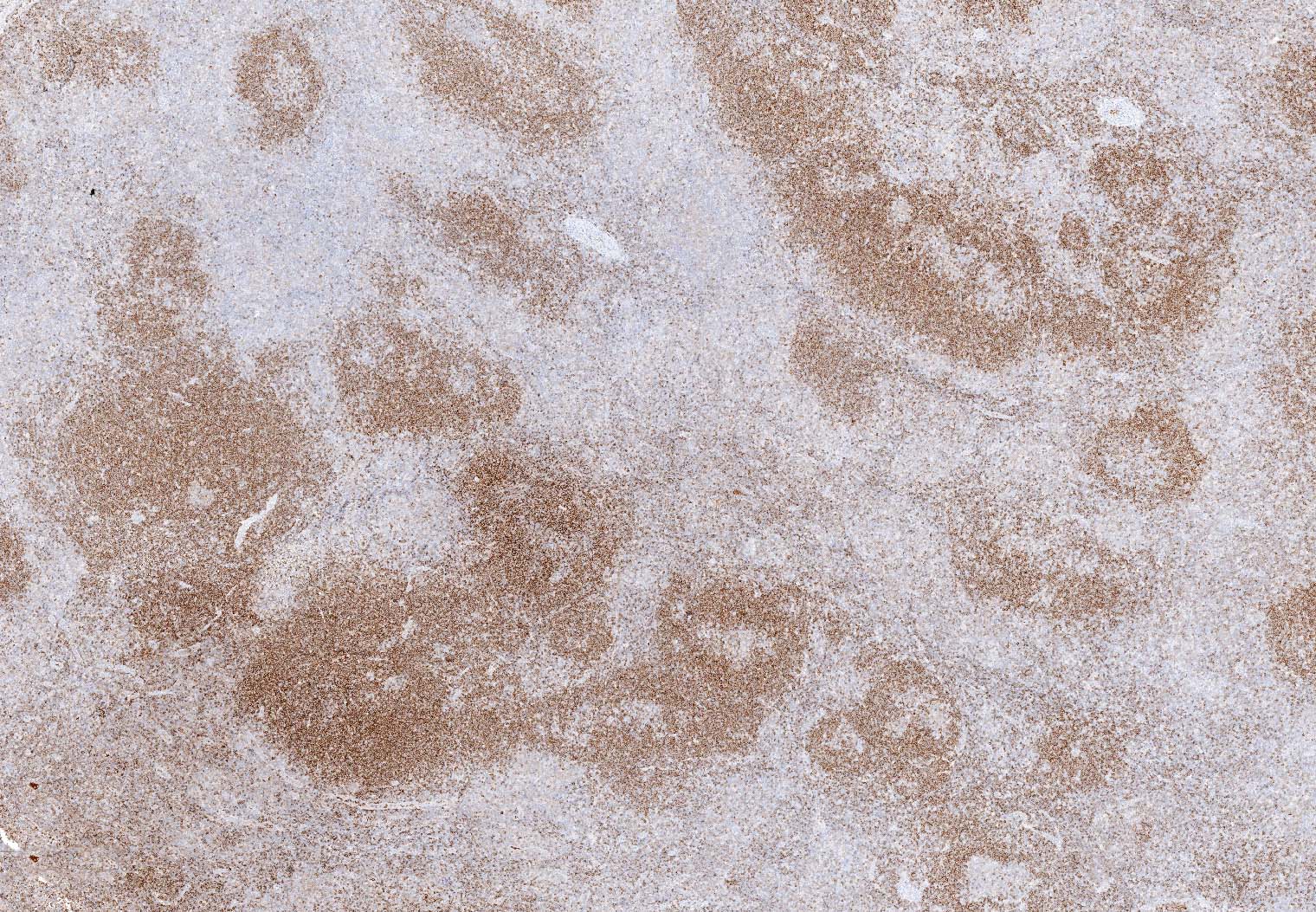
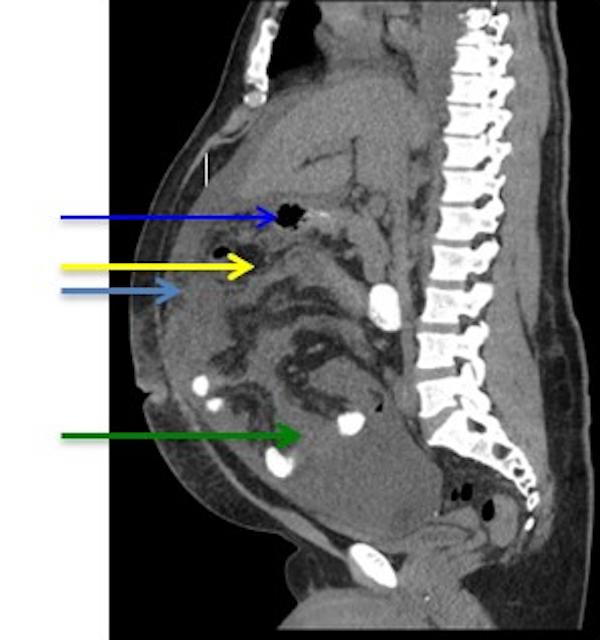
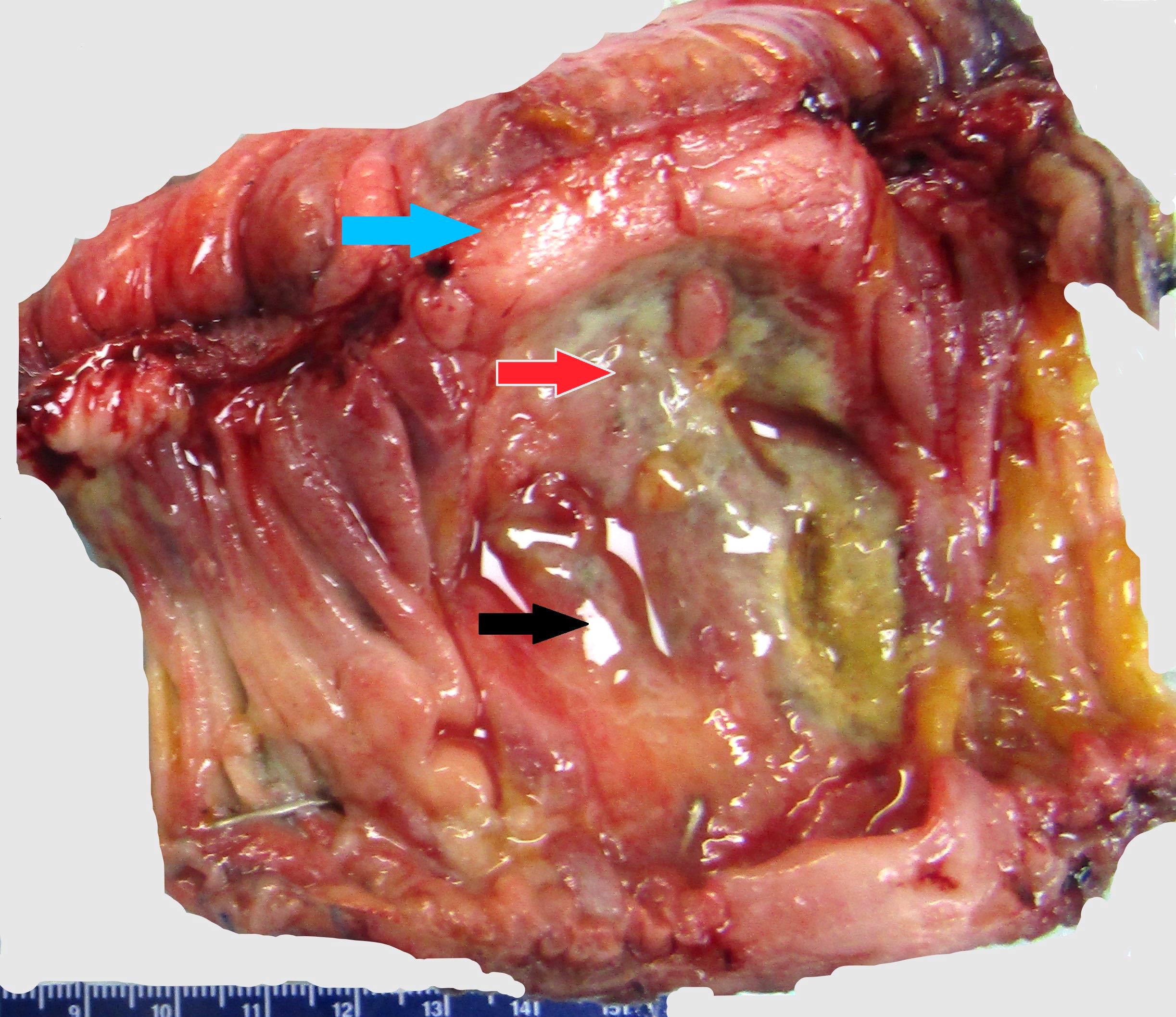
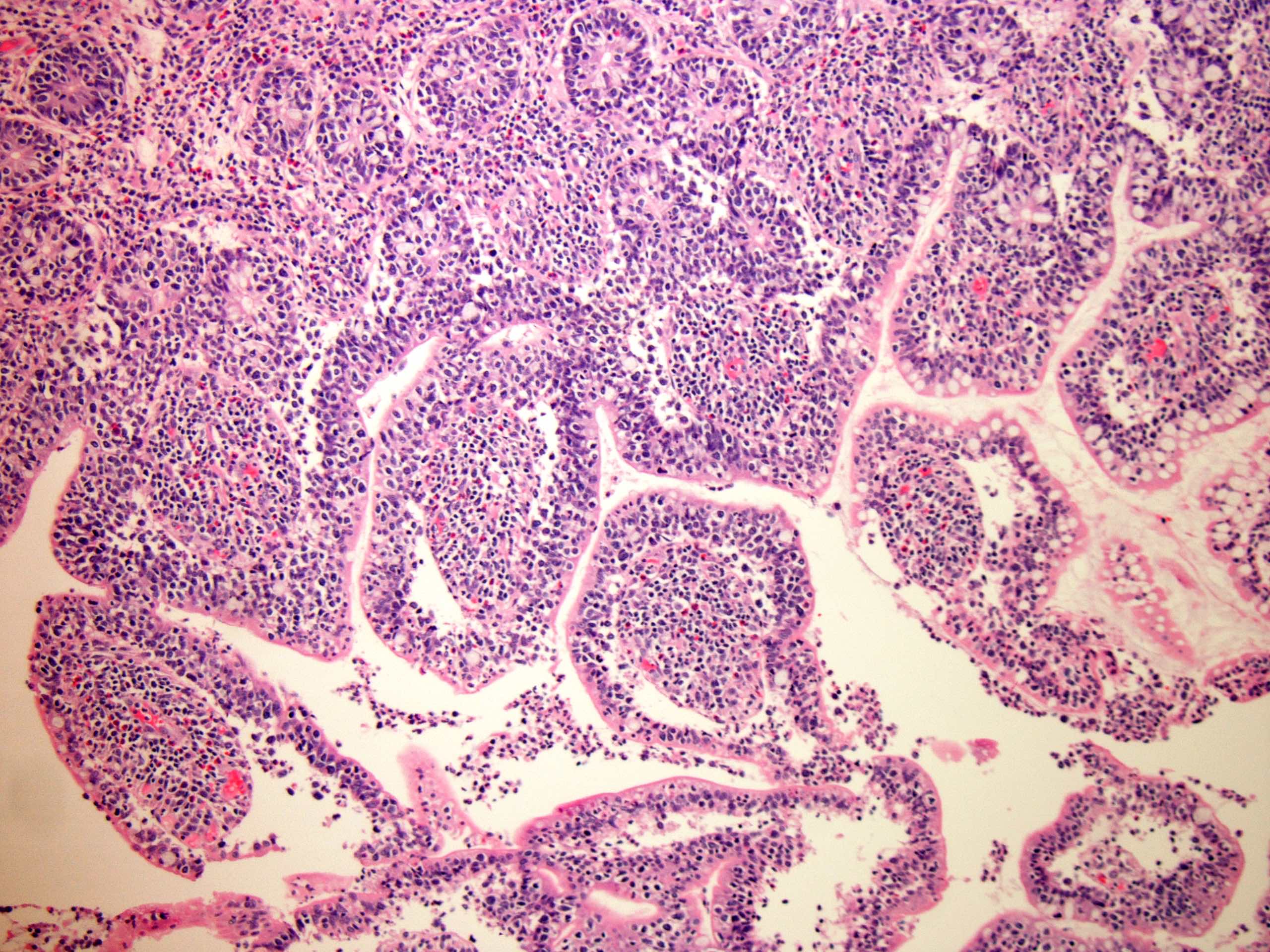
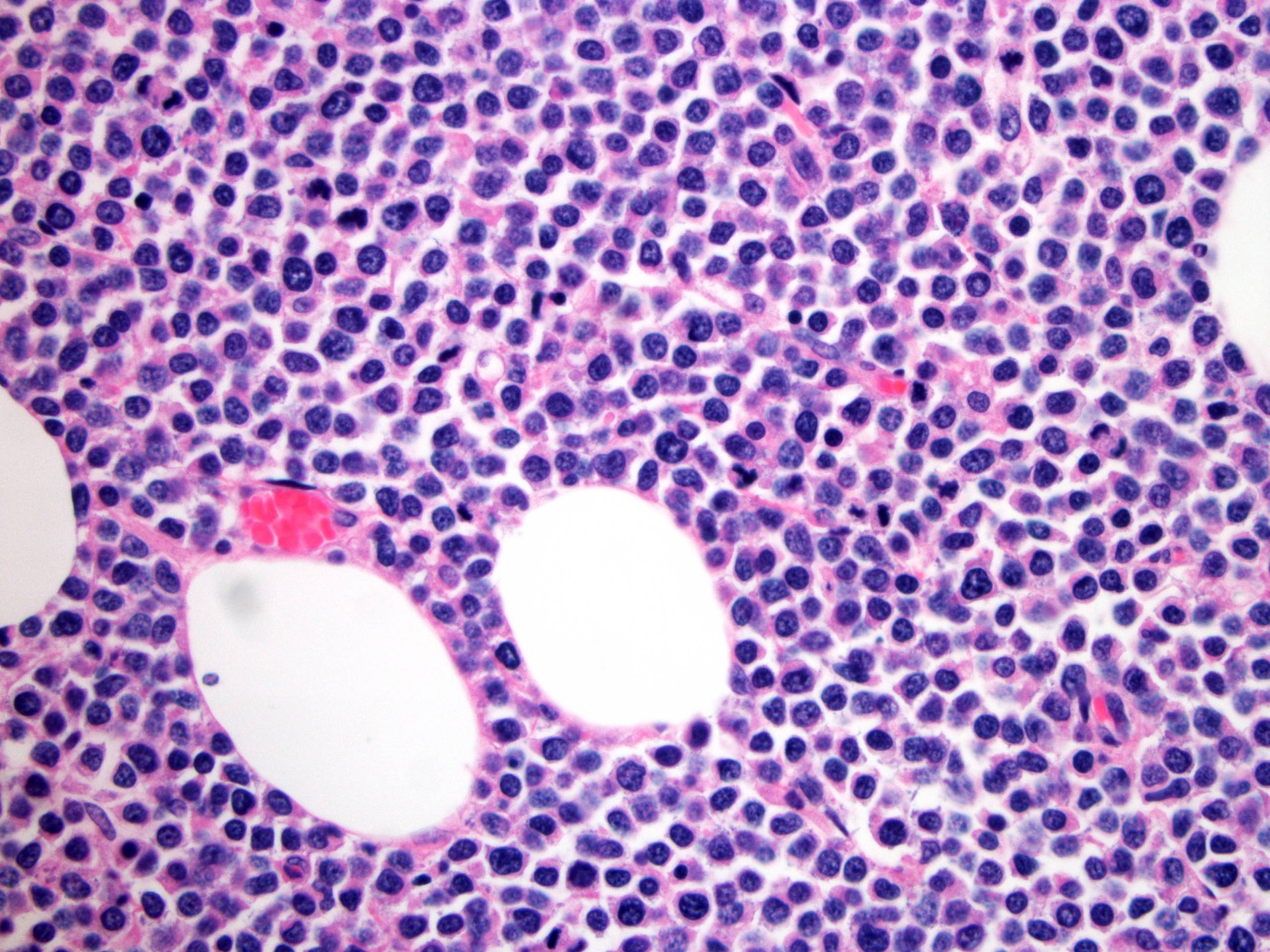
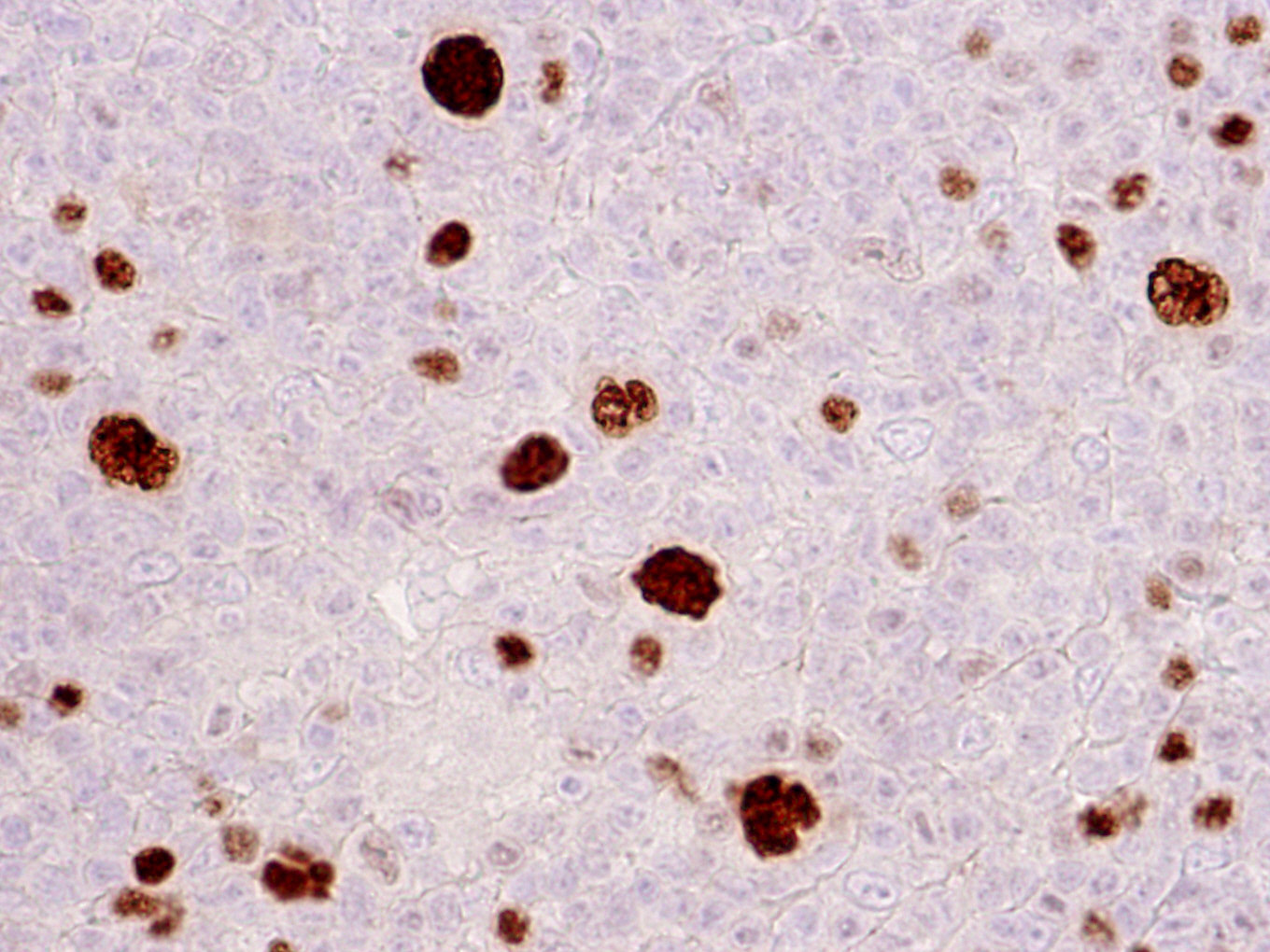
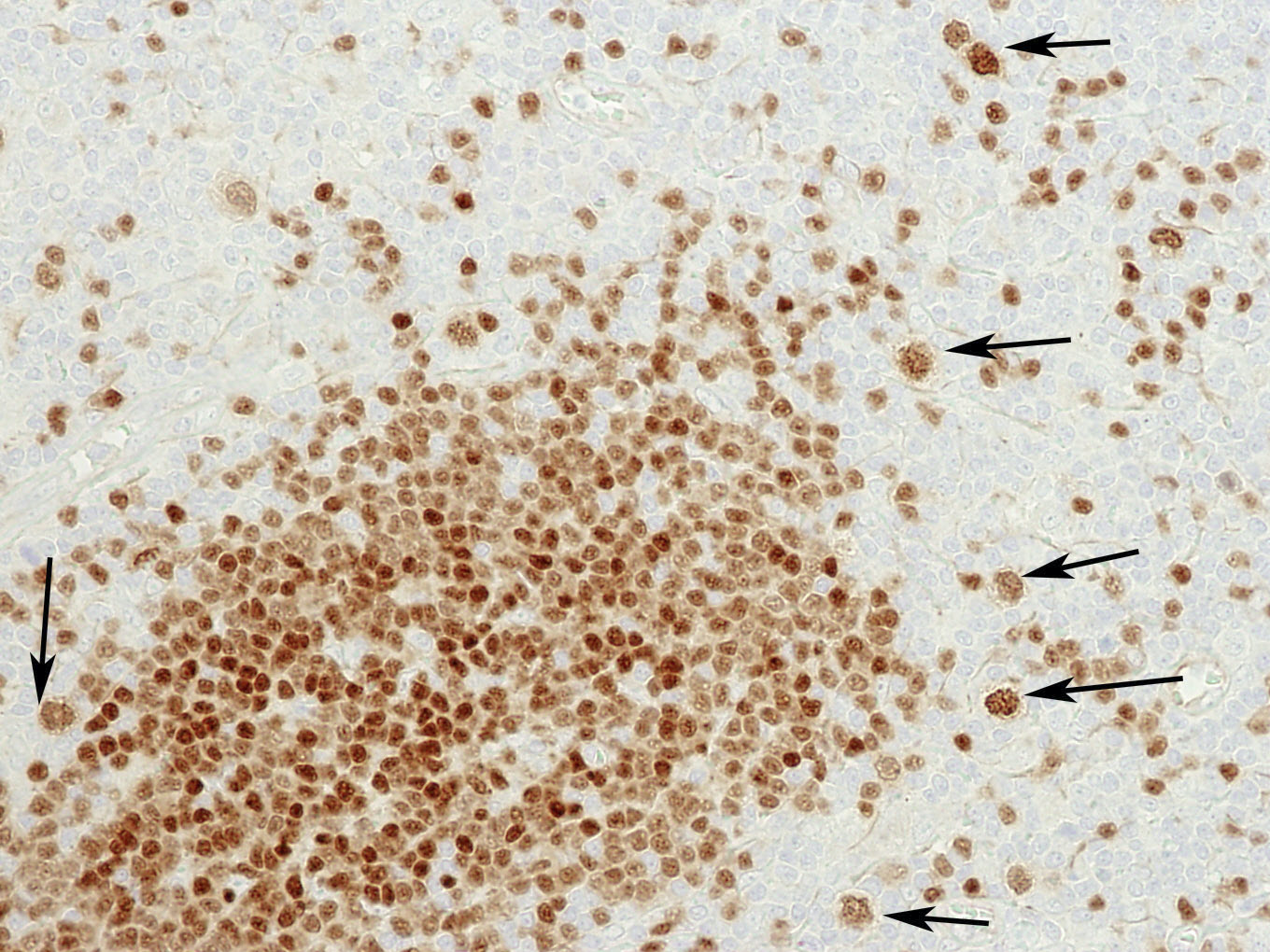
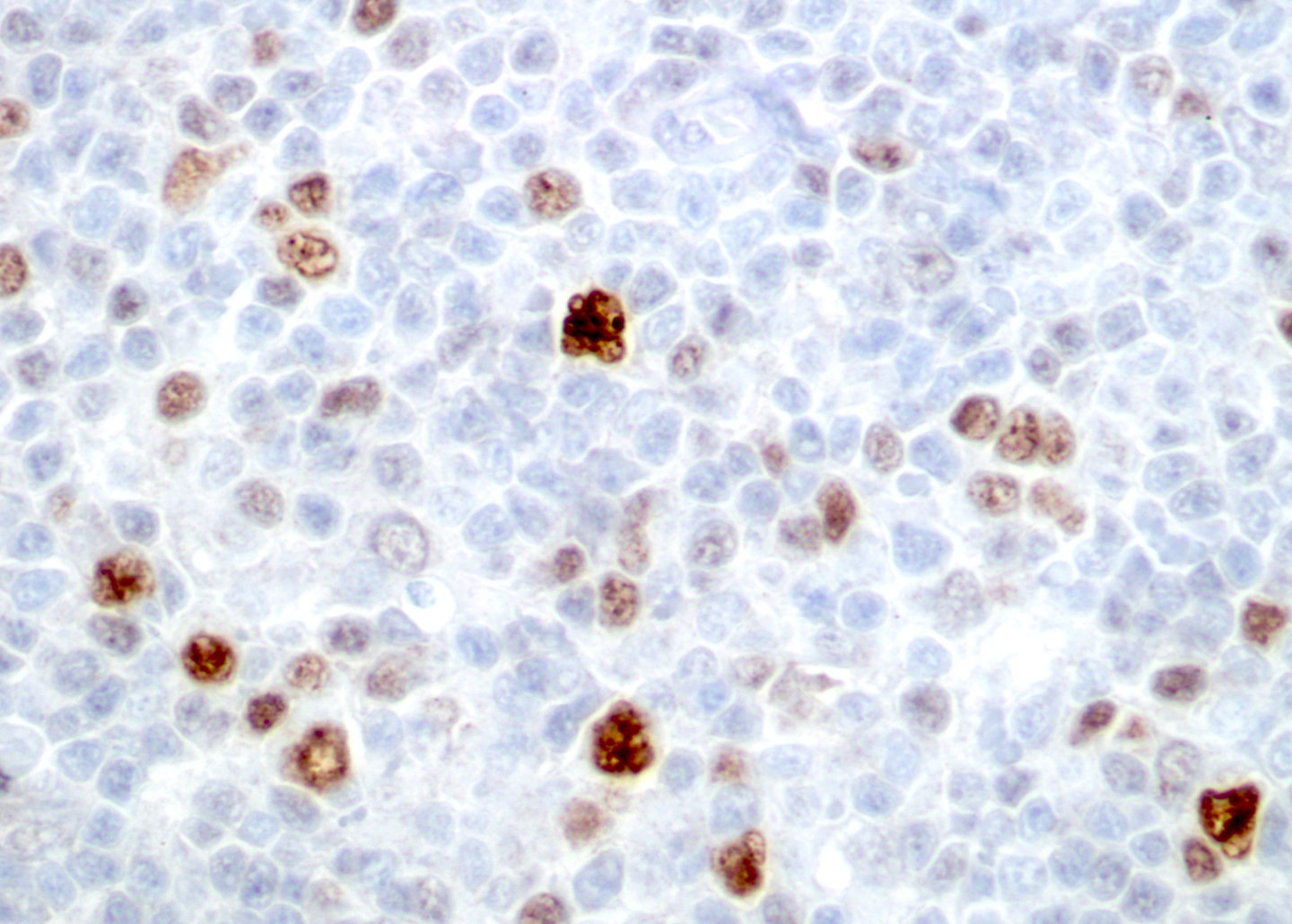
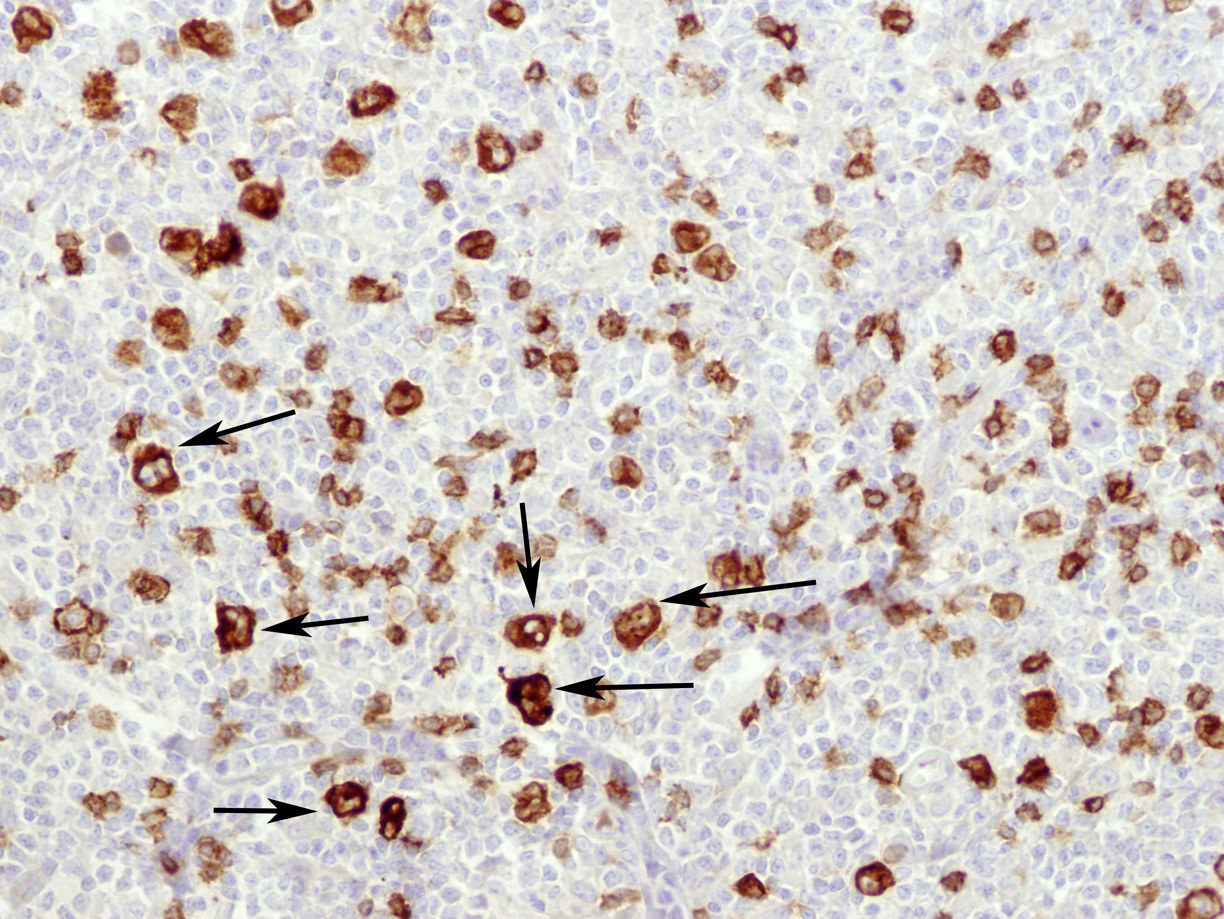
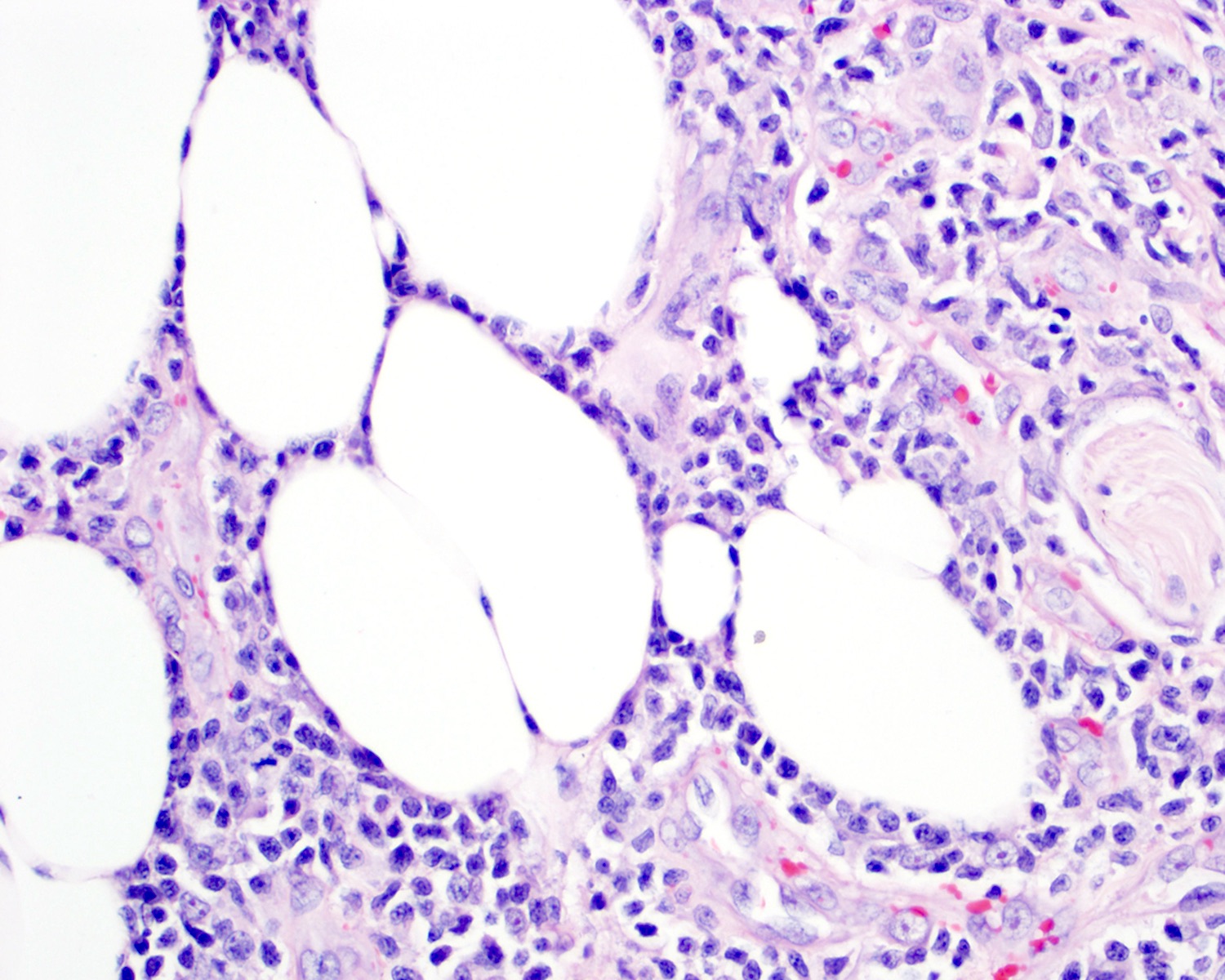
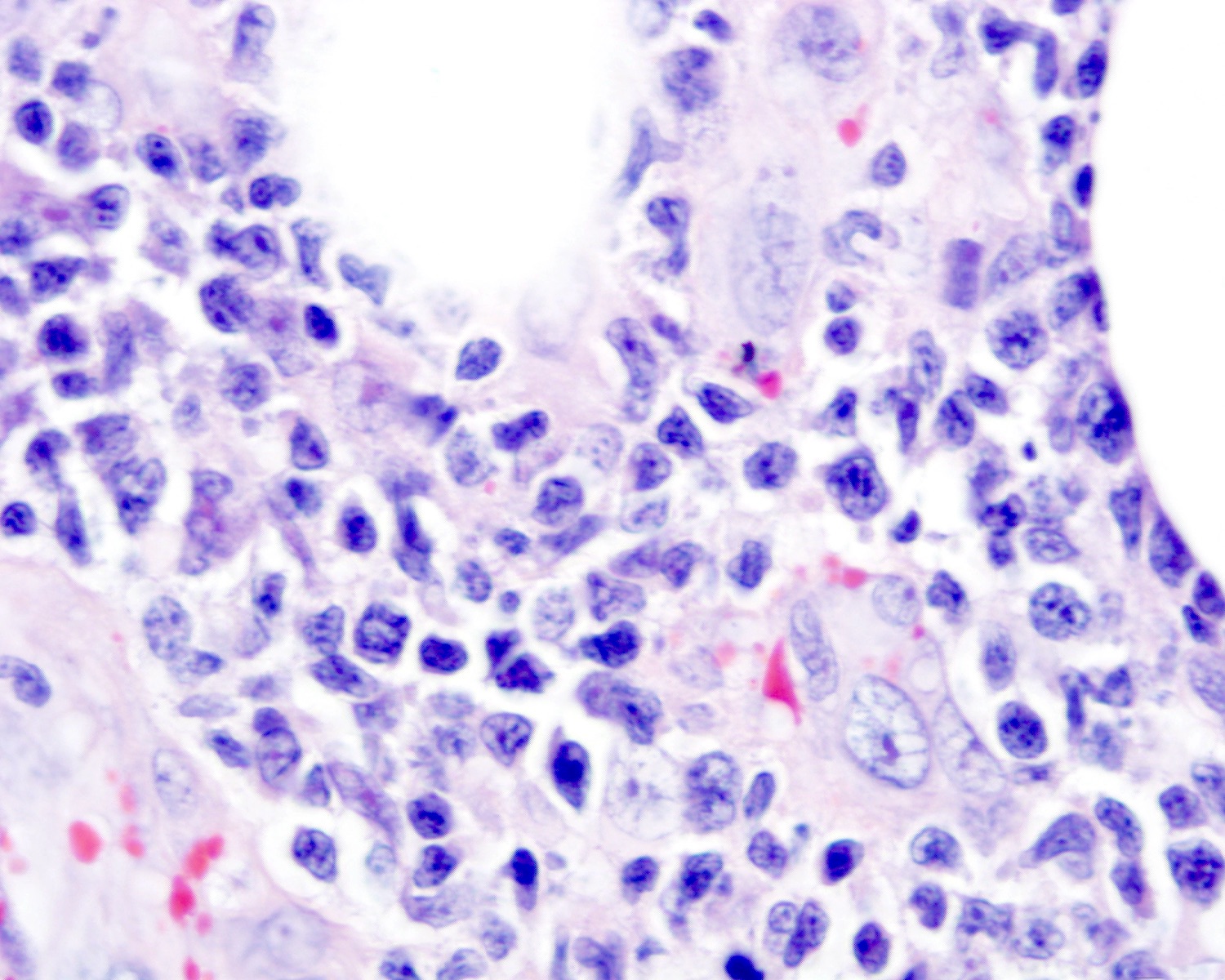
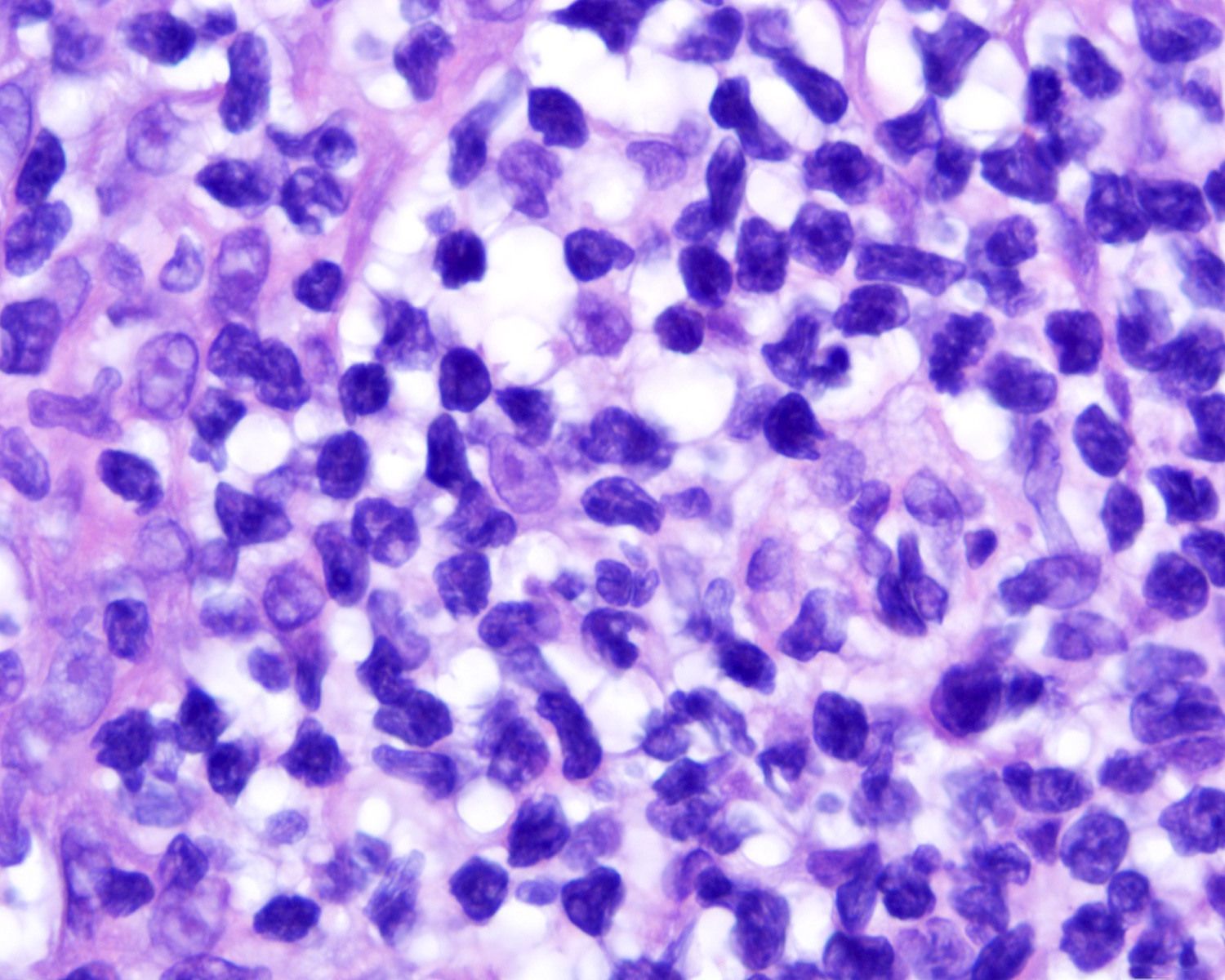
| WHO HAEM4R
| WHO HAEM5
| ICC
|
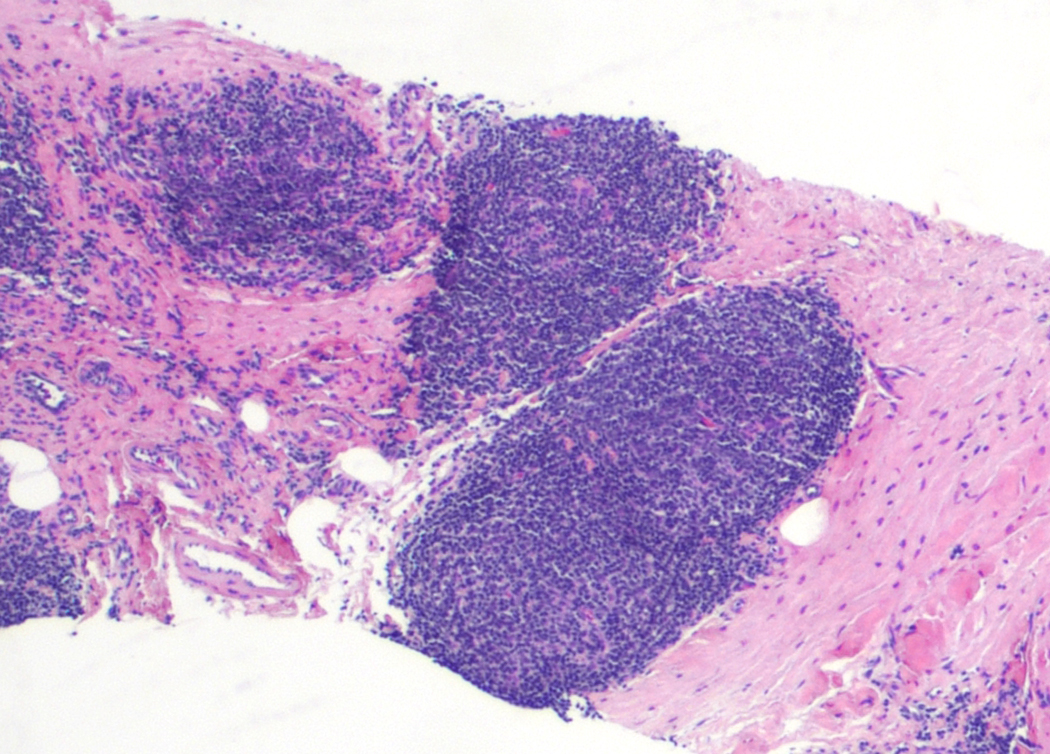
| Tumor-like lesions with B cell proliferation
|
| Not included
| Reactive B cell rich lymphoid proliferations that can mimic lymphoma
|
|
| Not included
| IgG4 related disease
|
|
| Not included
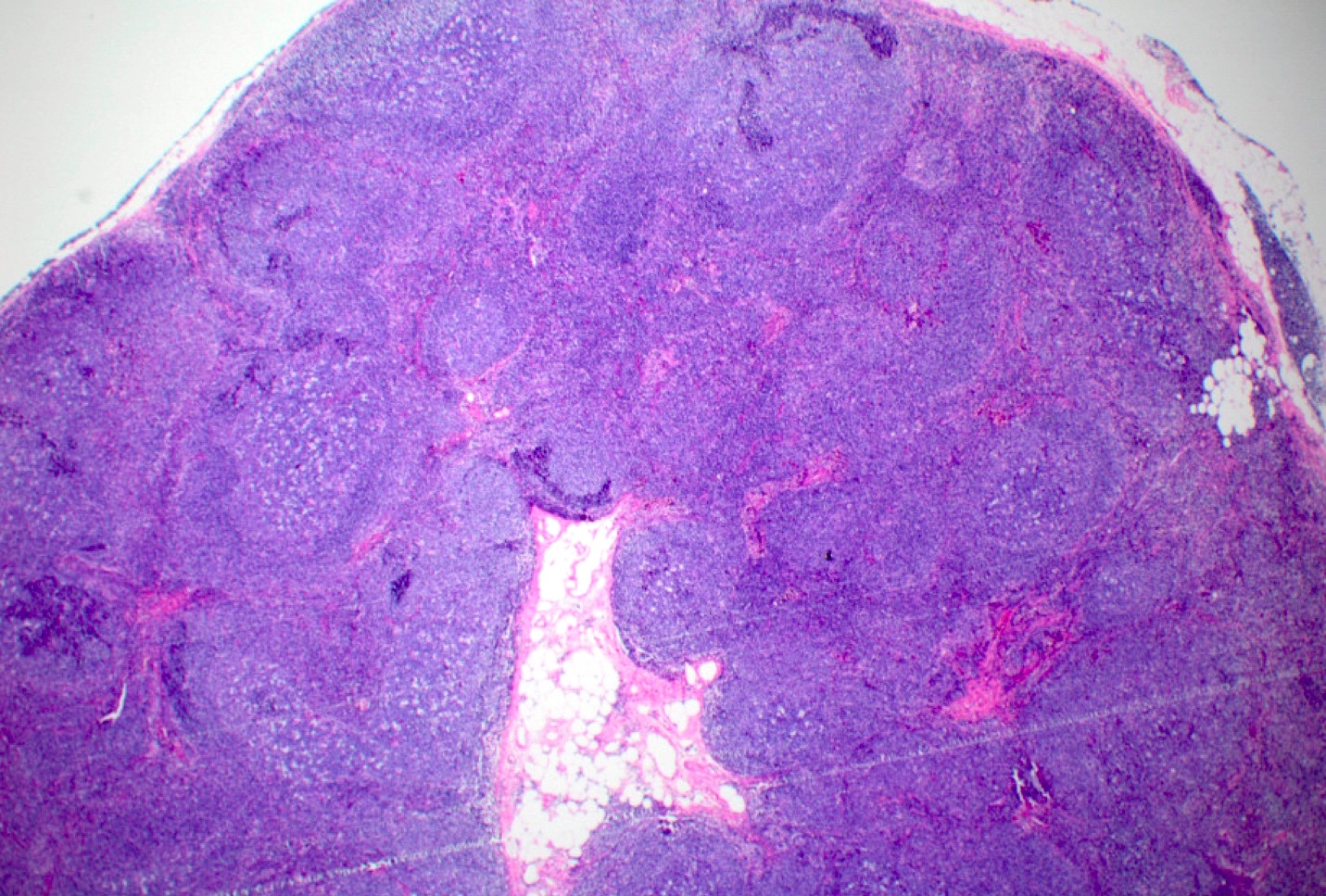
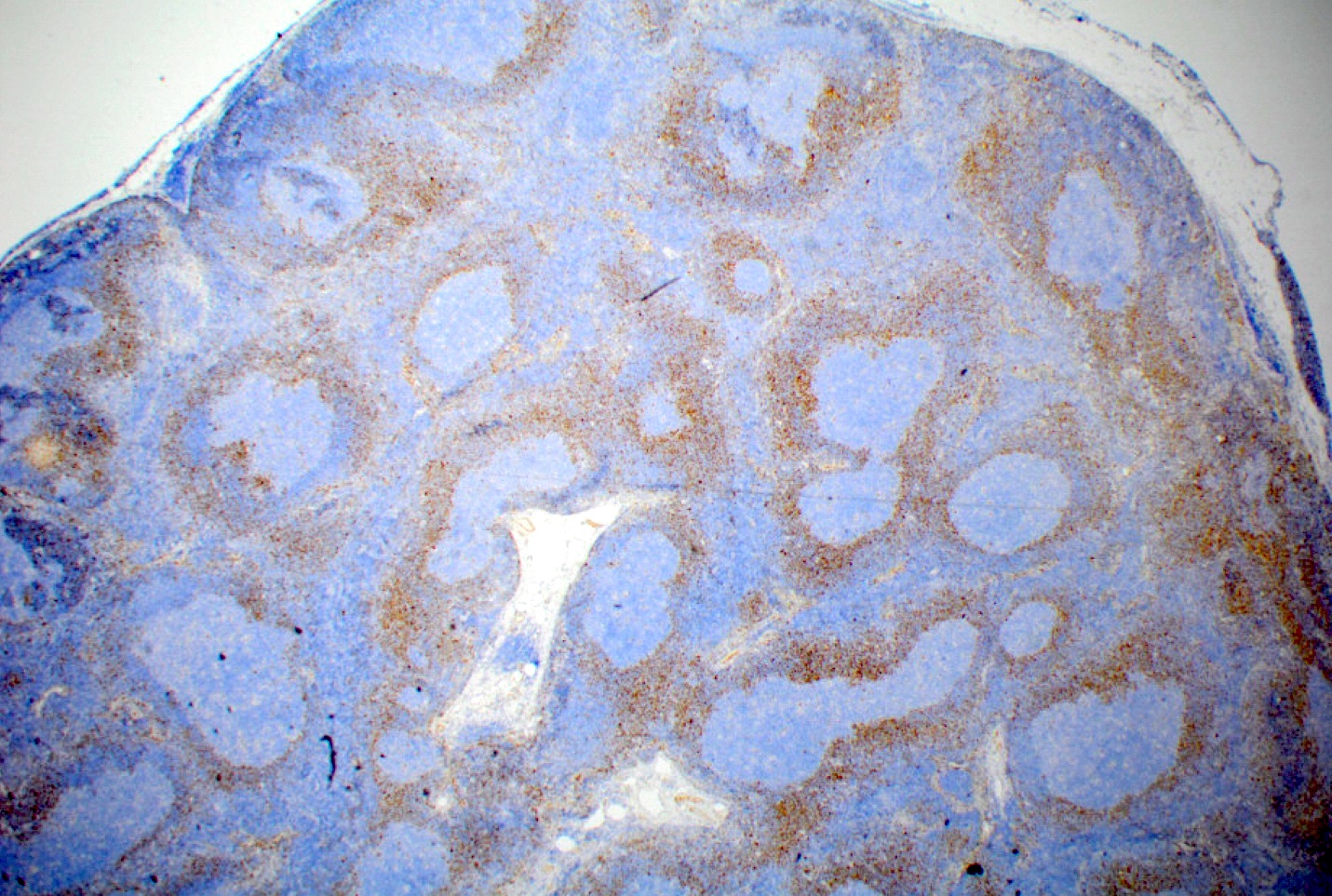
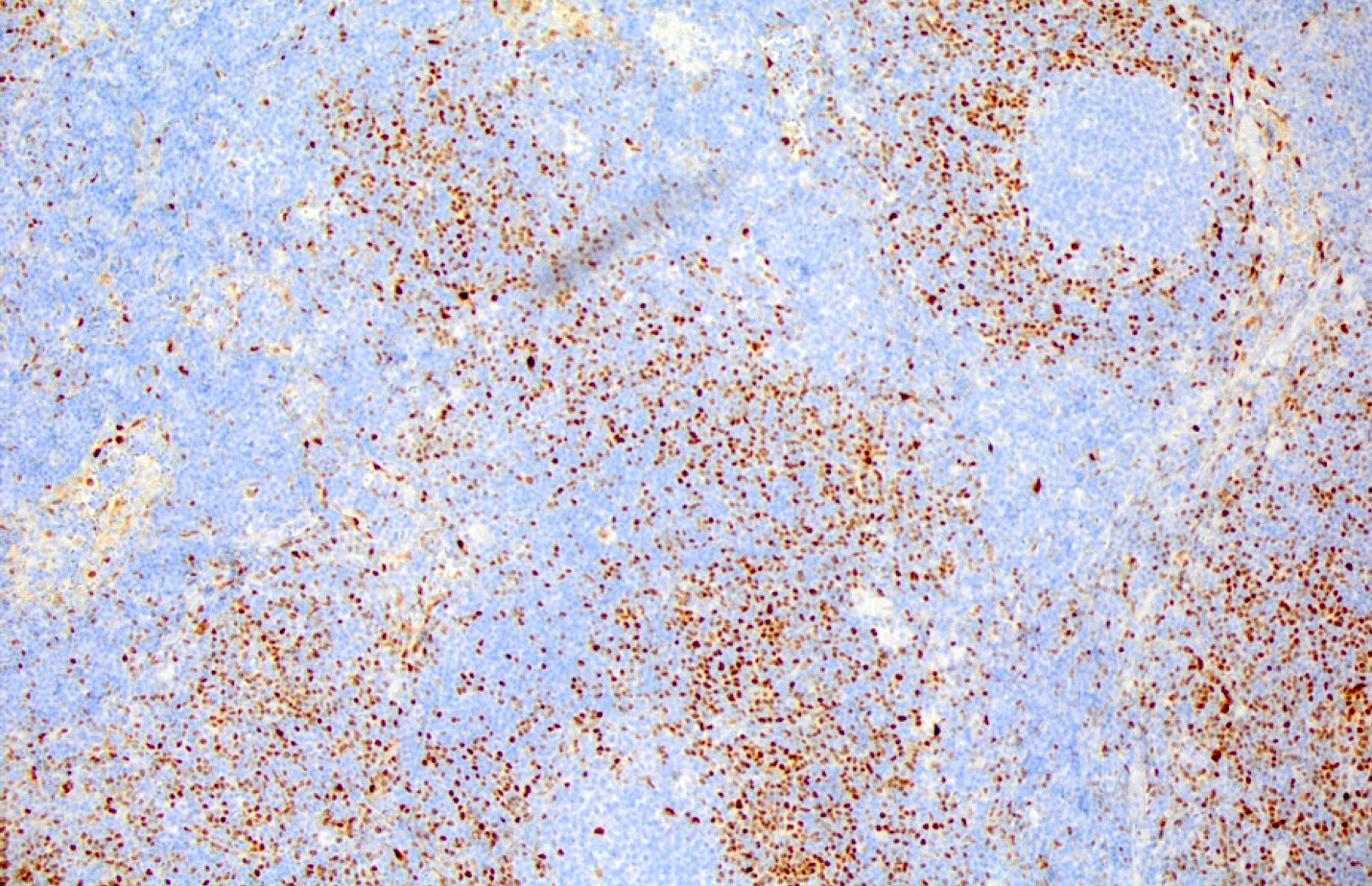
| Unicentric Castleman disease
|
|
| Not included
| Idiopathic multicentric Castleman disease
|
|
| Multicentric Castleman disease
| KSHV / HHV8 associated multicentric Castleman disease
| Multicentric Castleman disease
|
| Not included
|
| EBV positive polymorphic B cell lymphoproliferative disorder, NOS
|
| Neoplastic and preneoplastic small lymphocytic proliferation
|
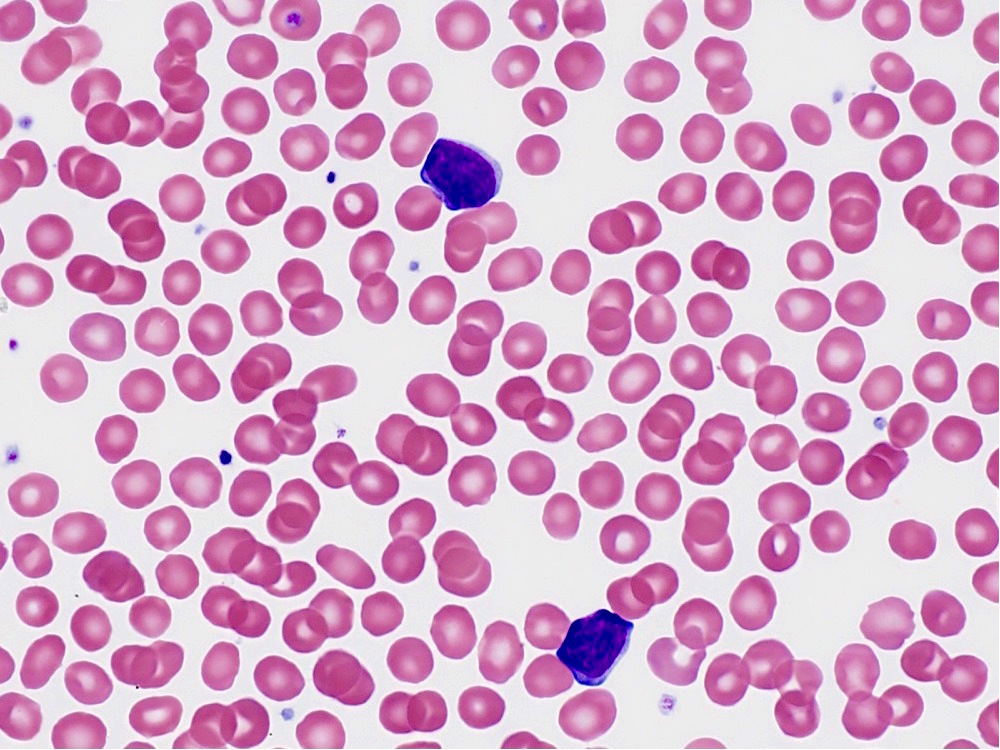
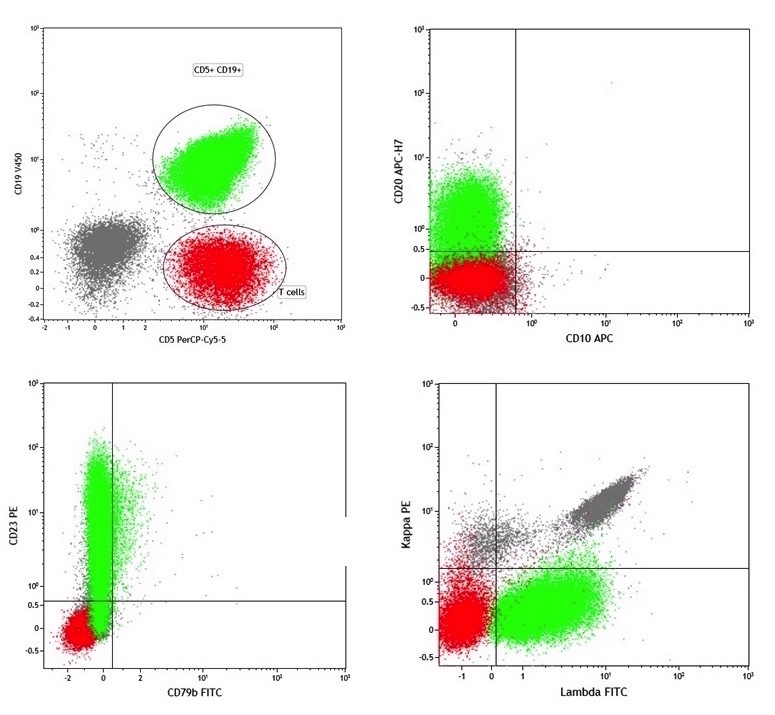
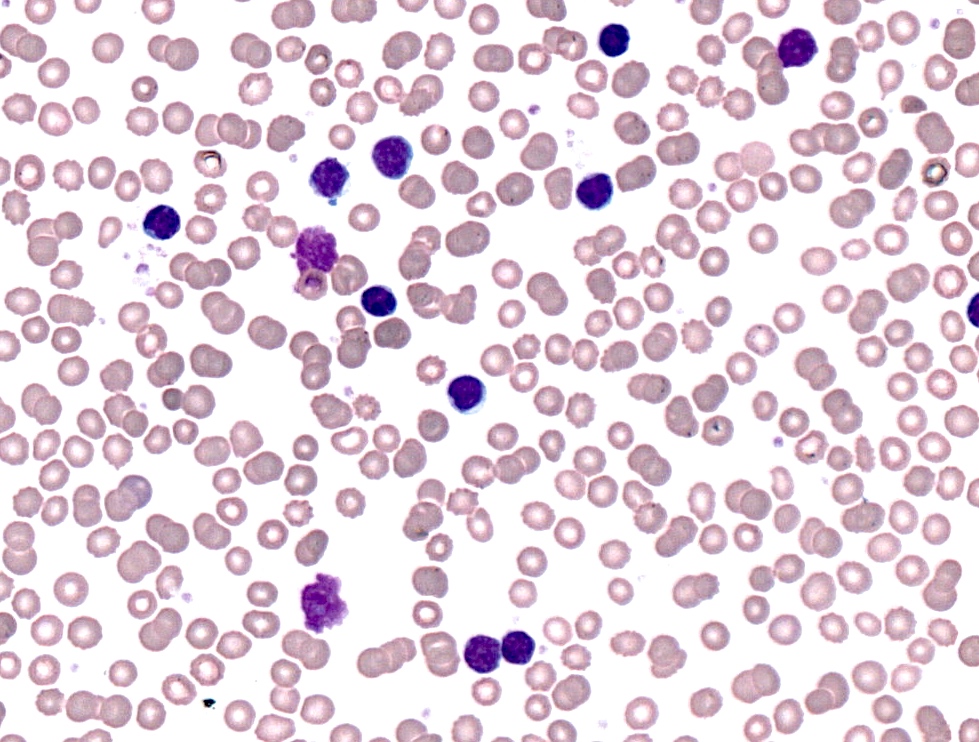
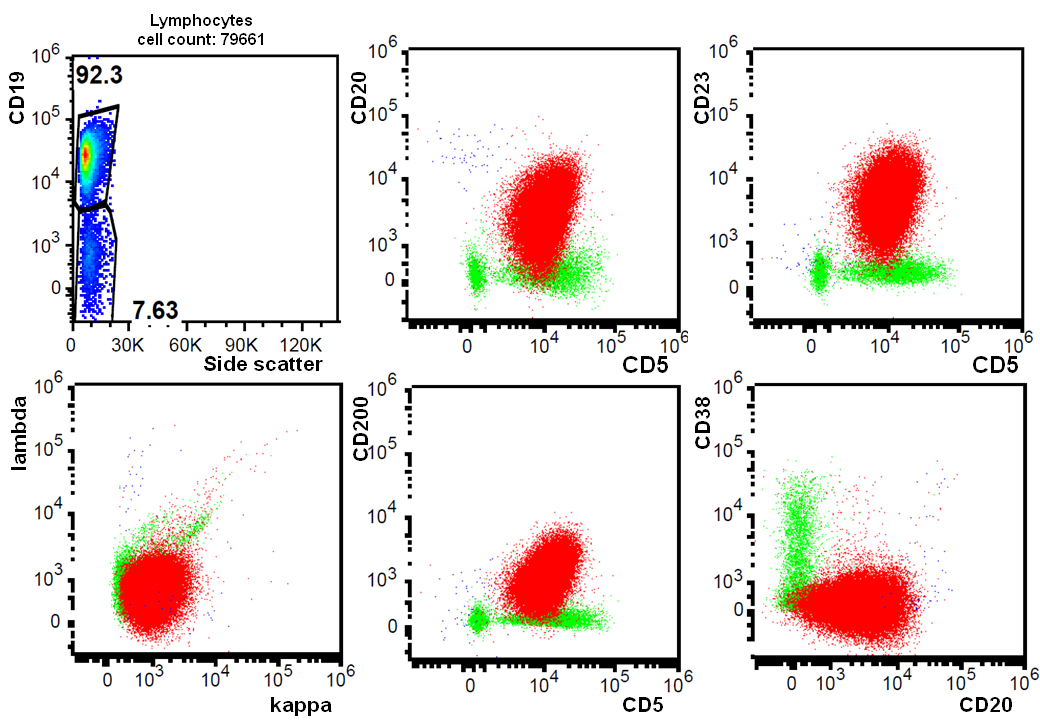
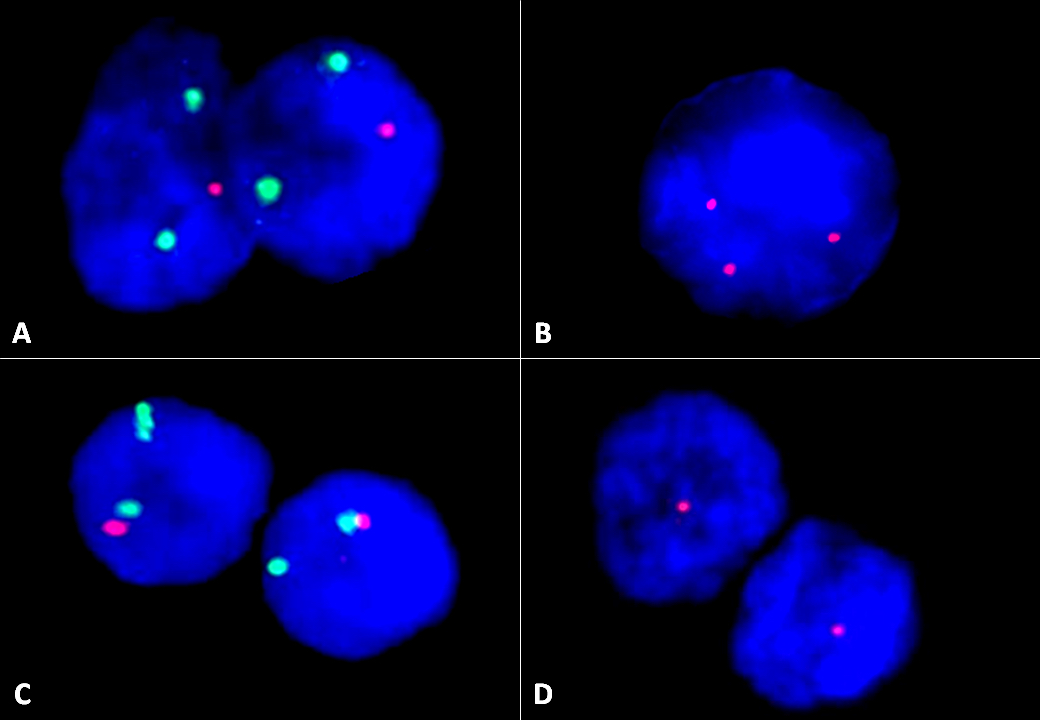
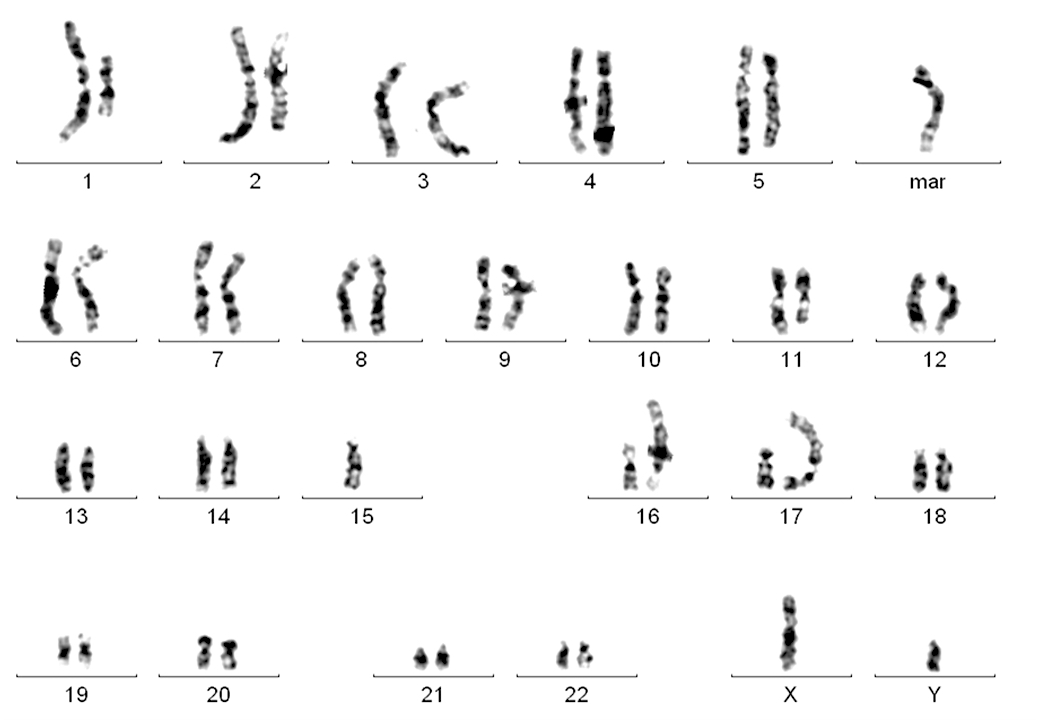
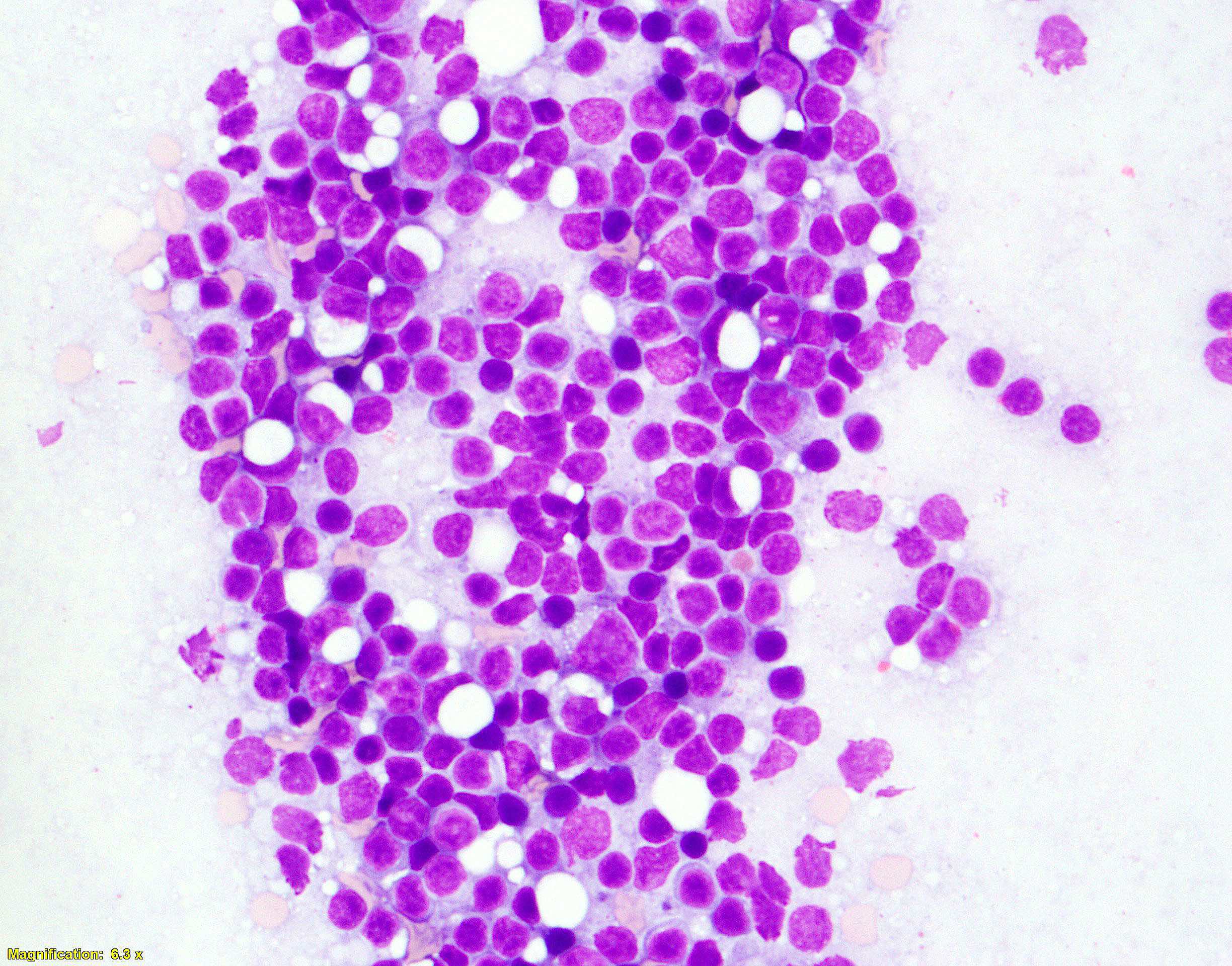
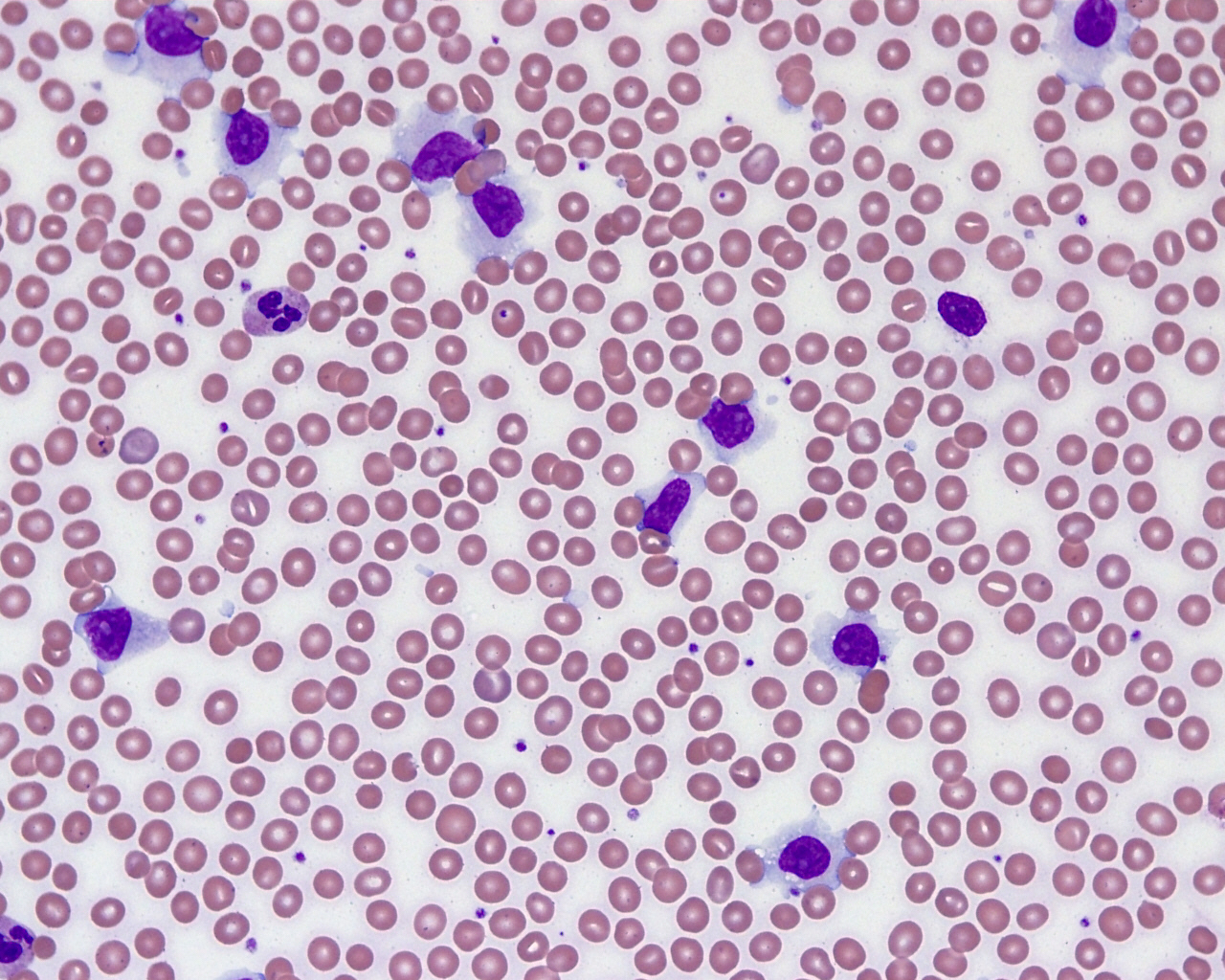
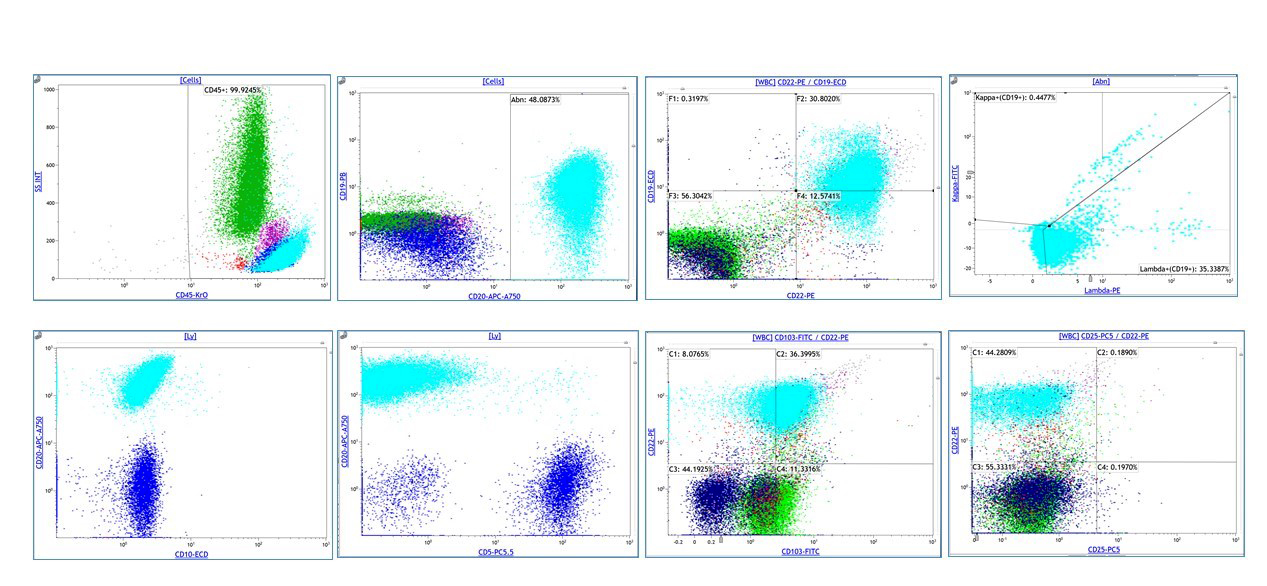
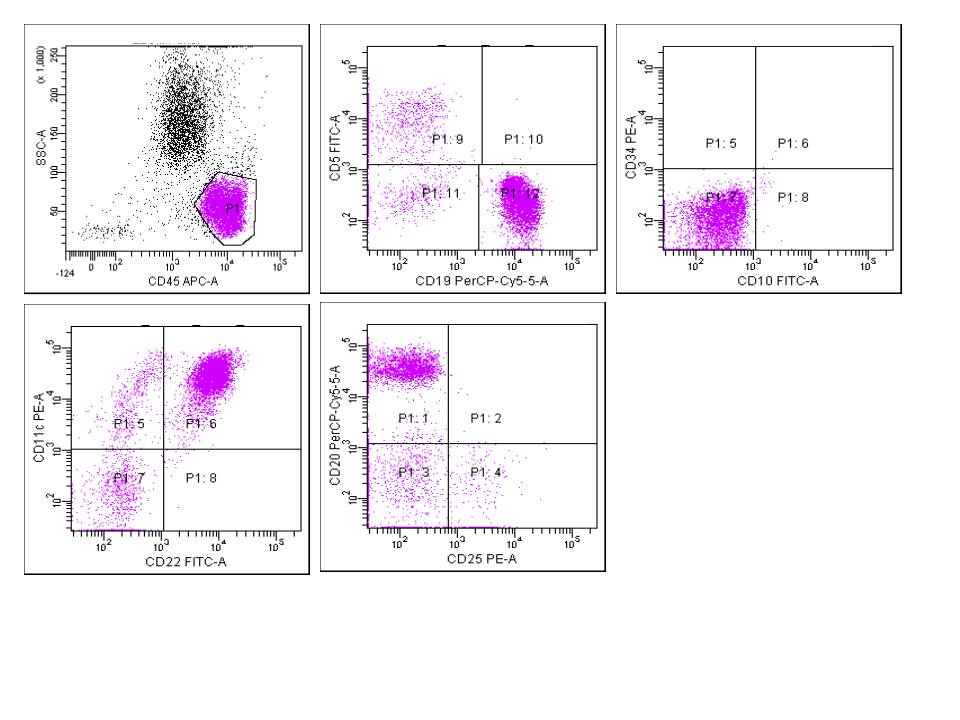
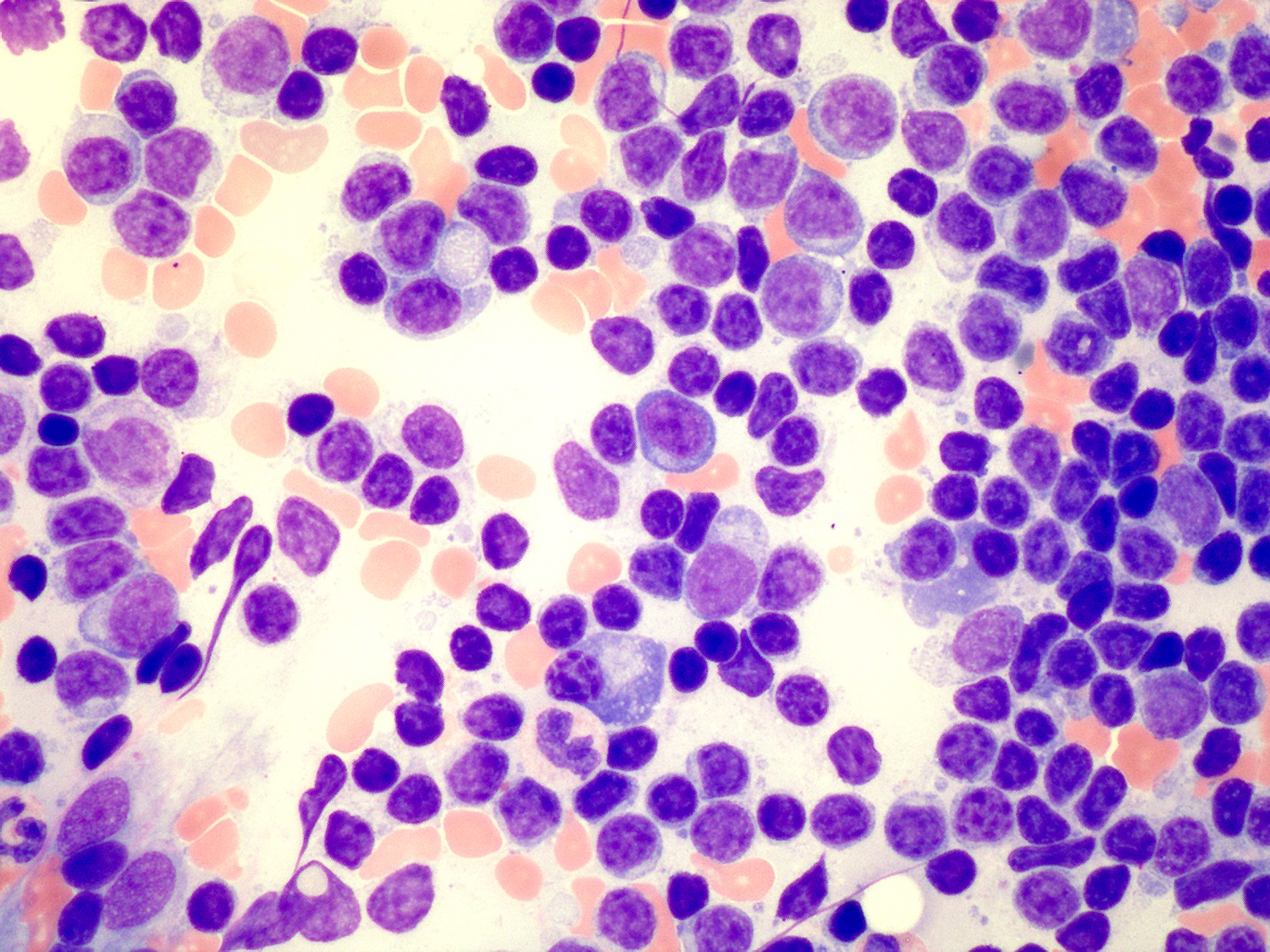
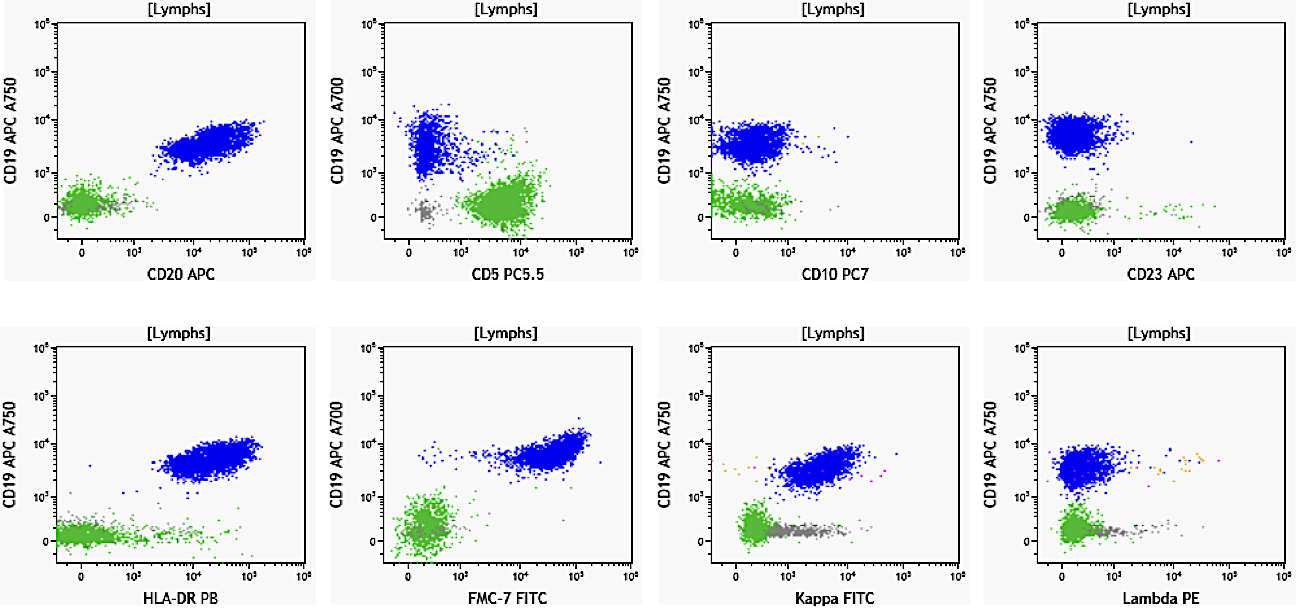
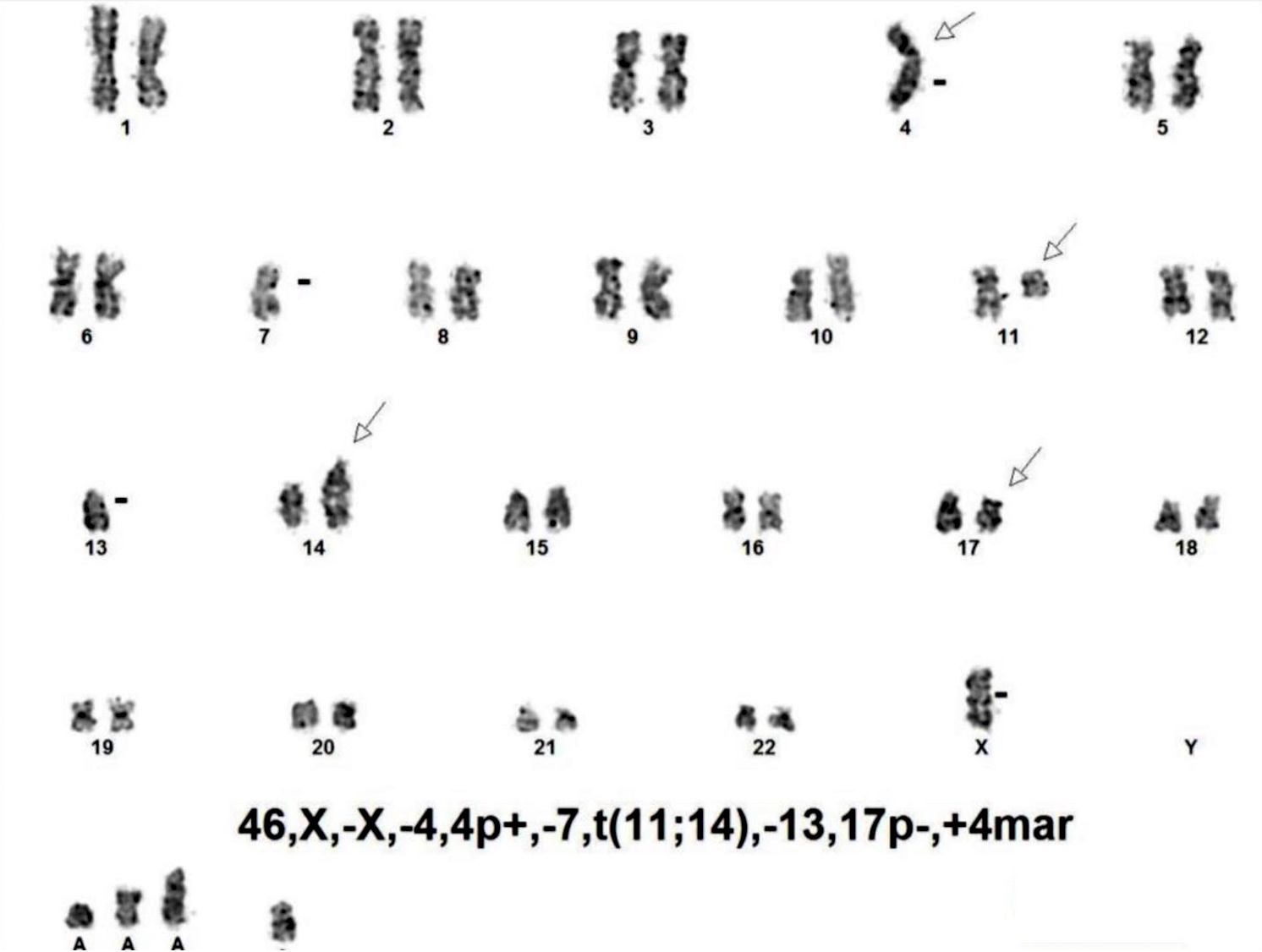
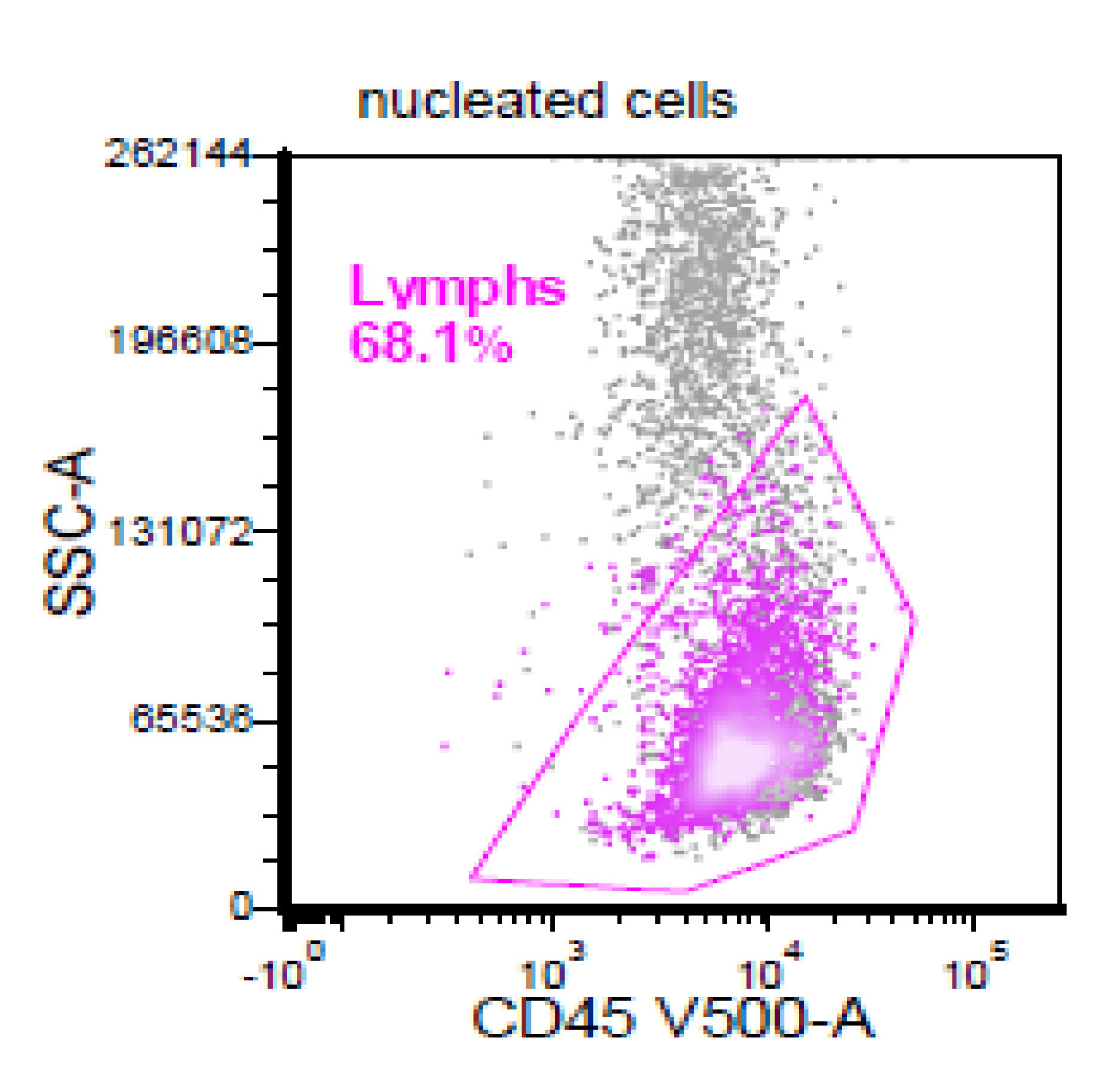
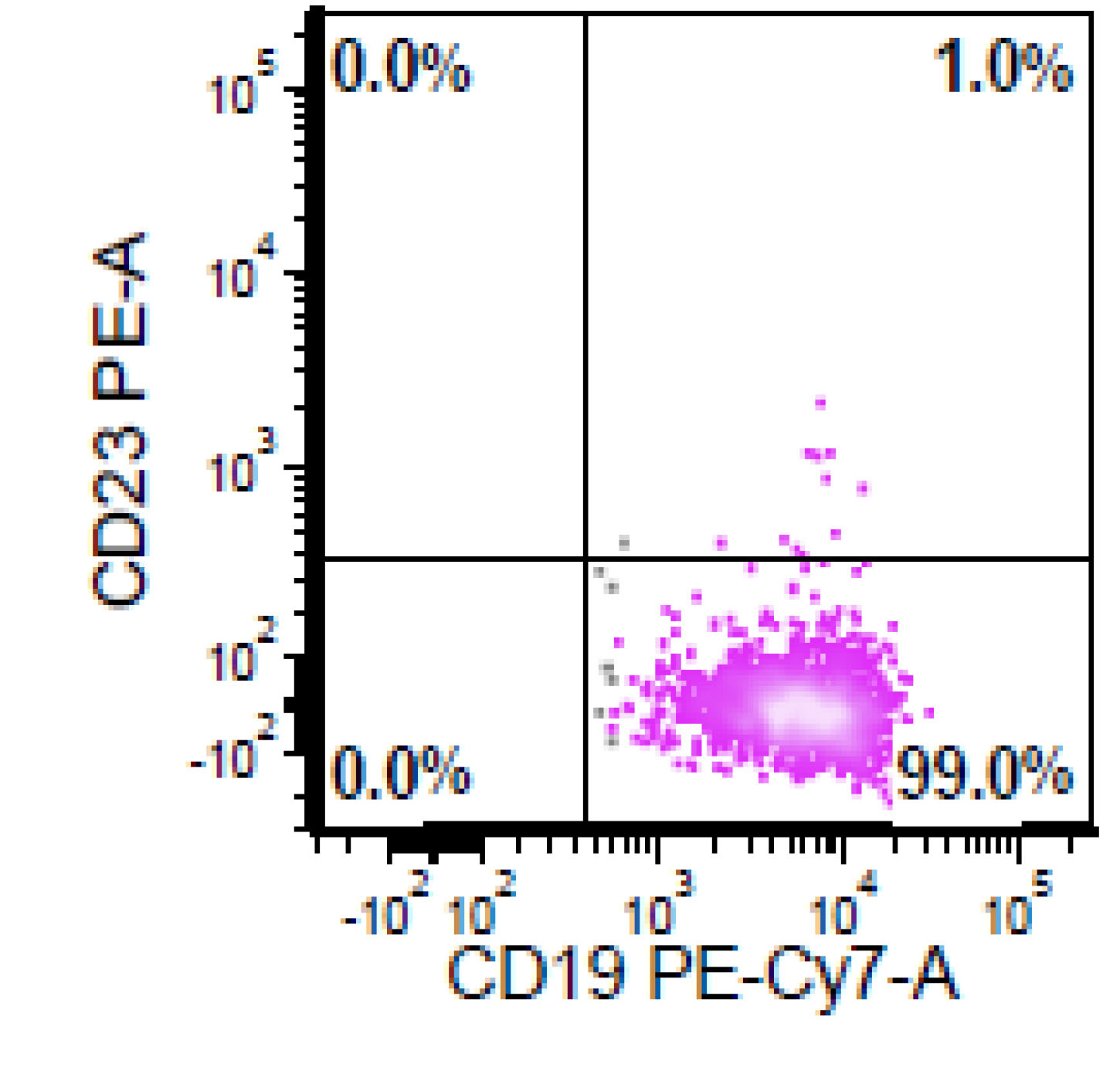
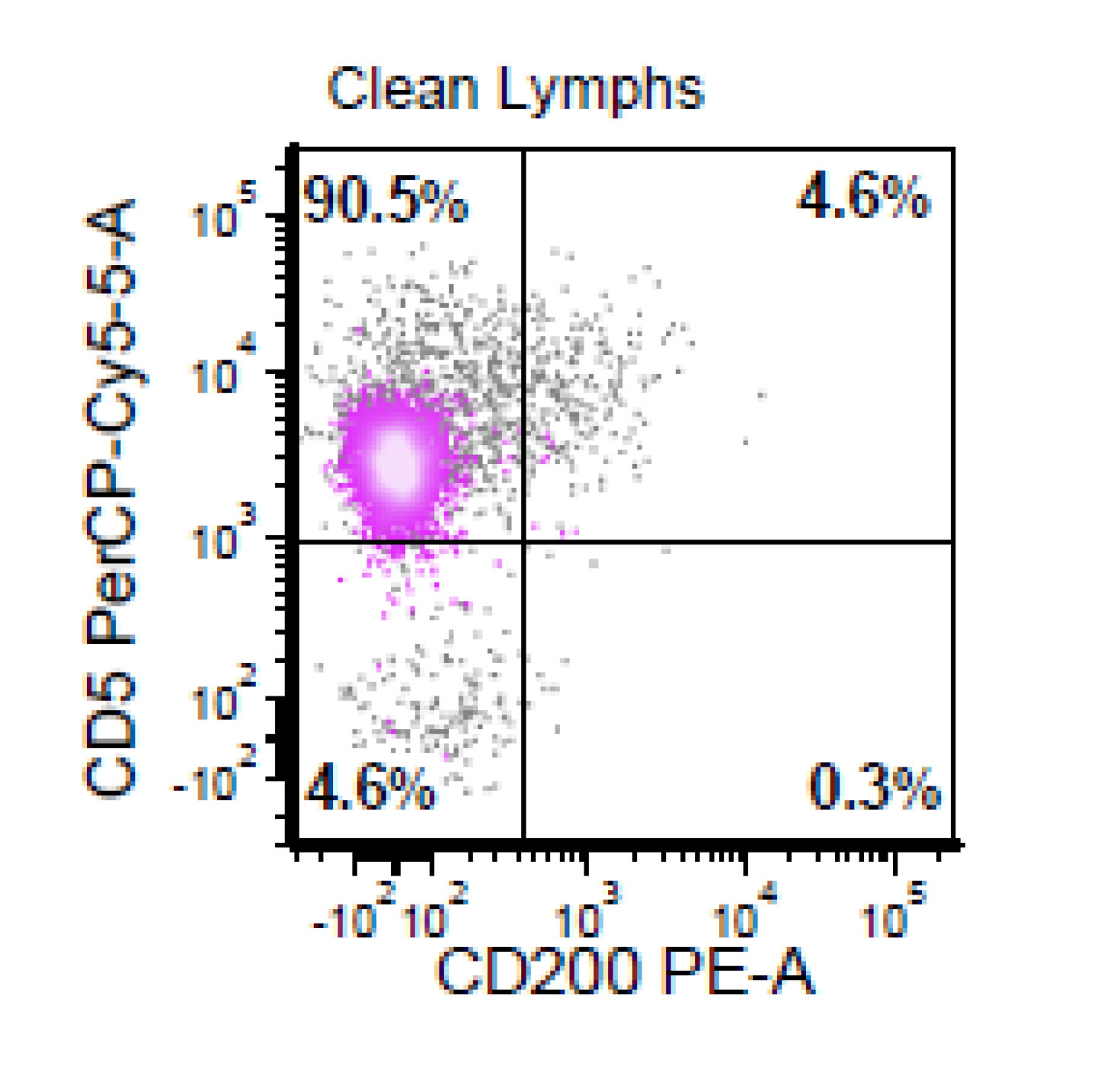
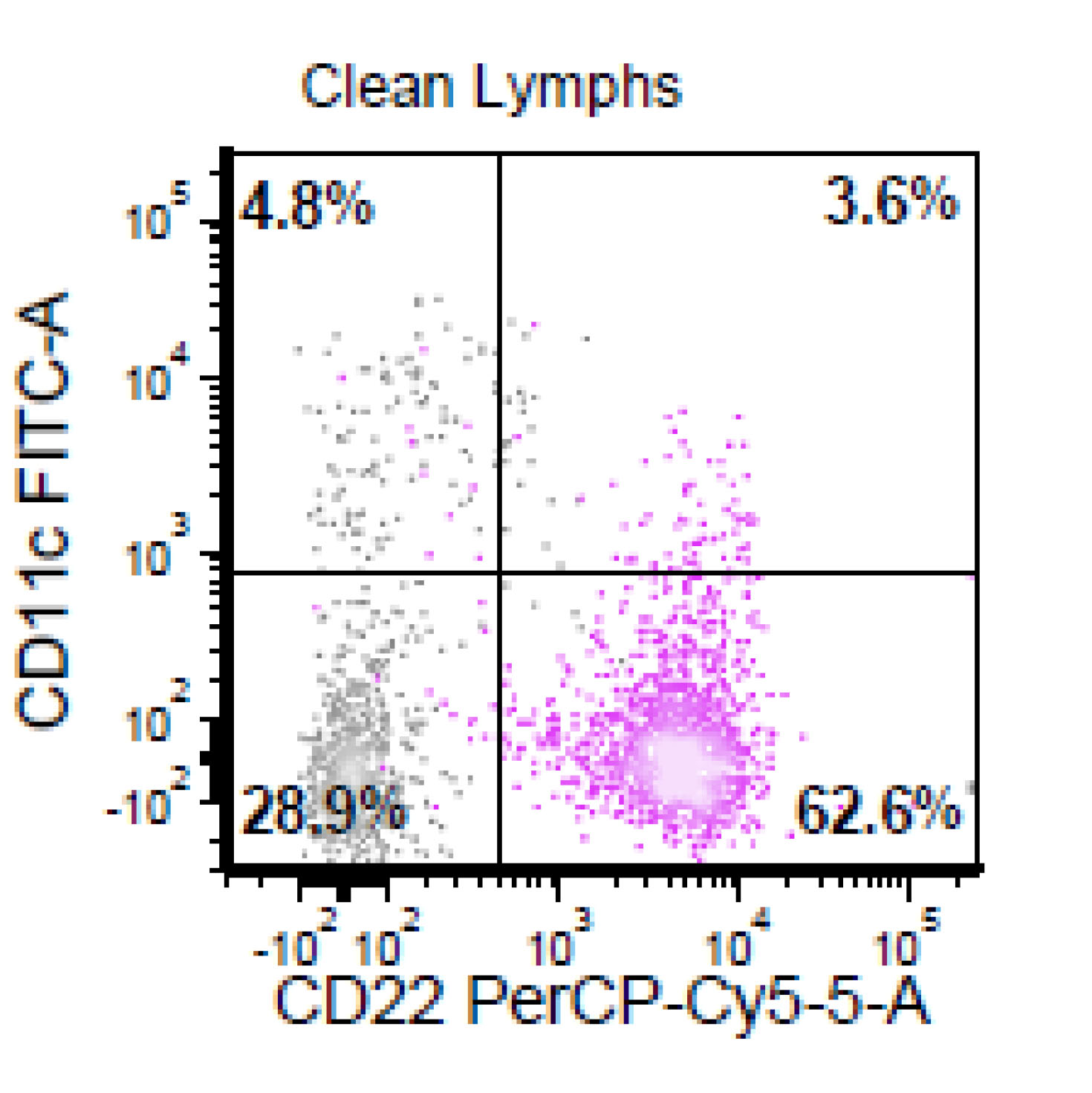
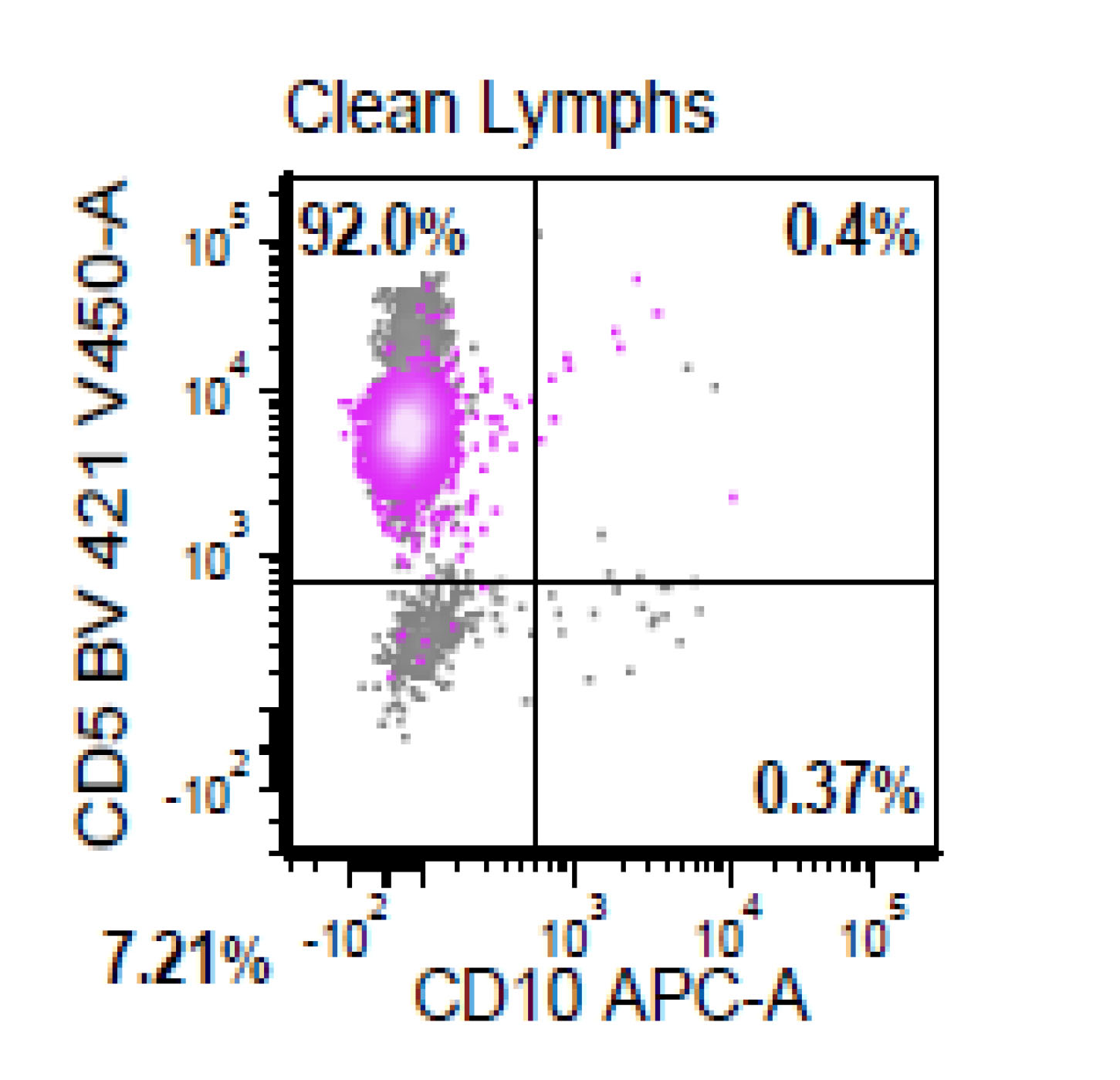
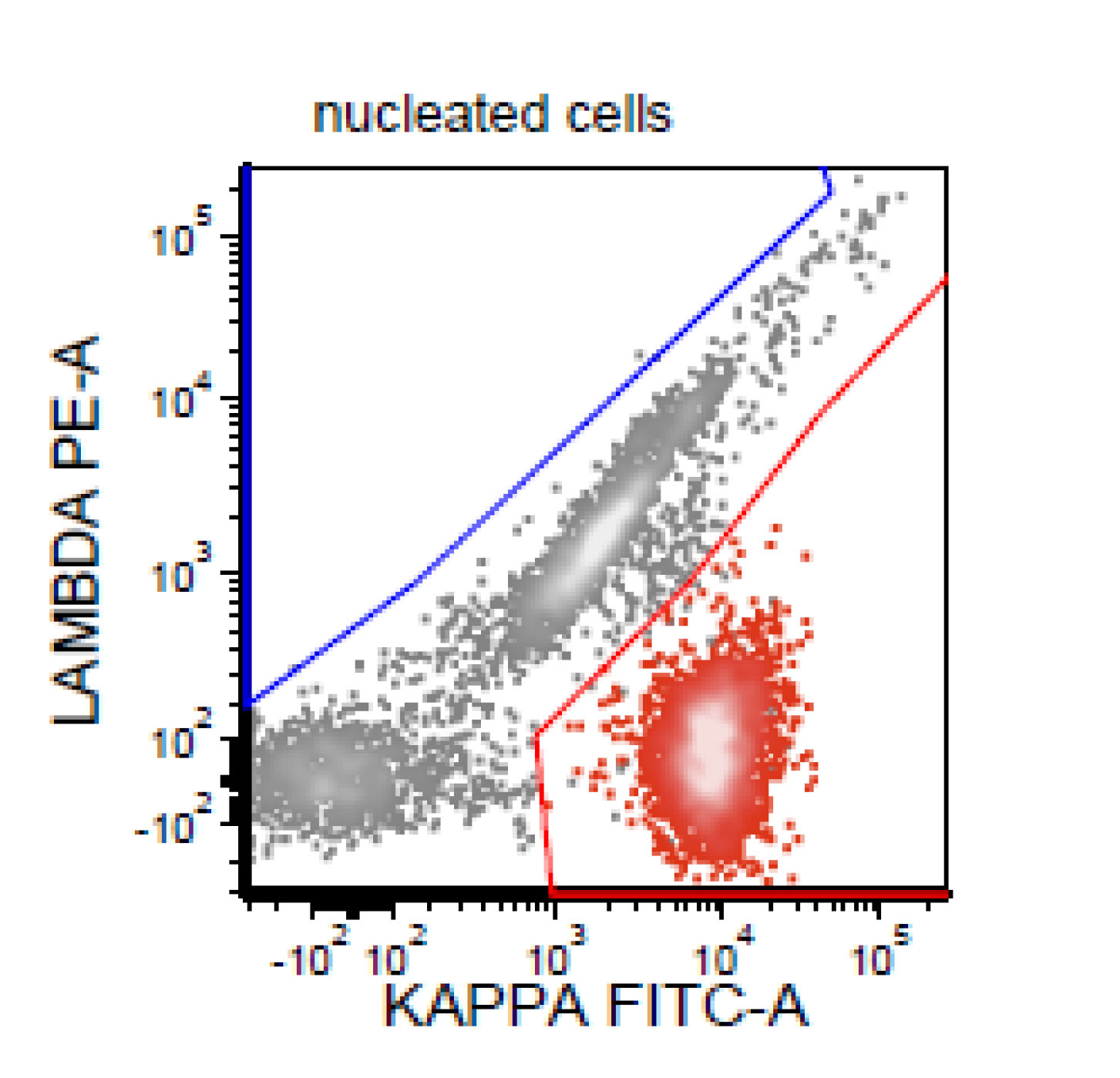
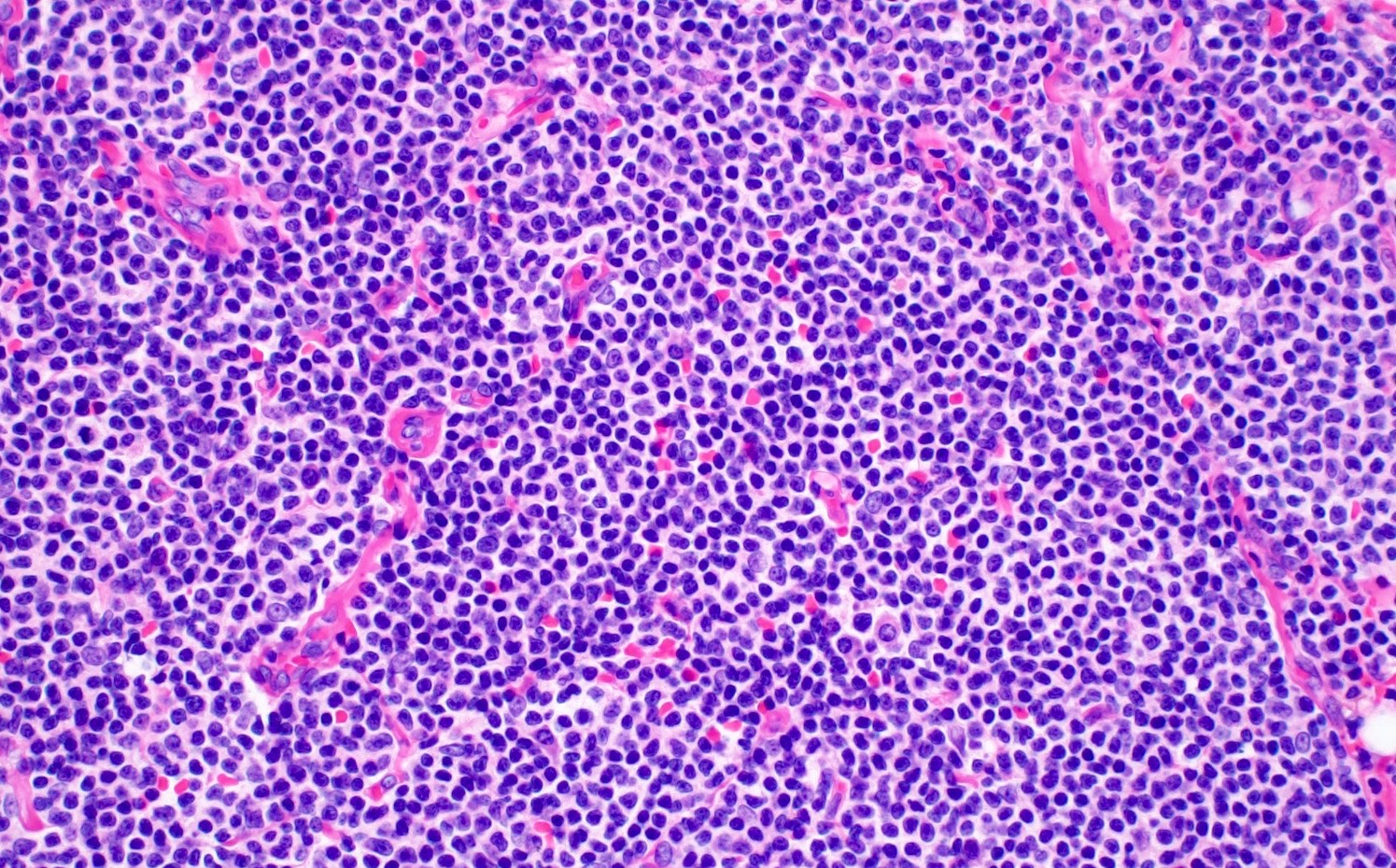
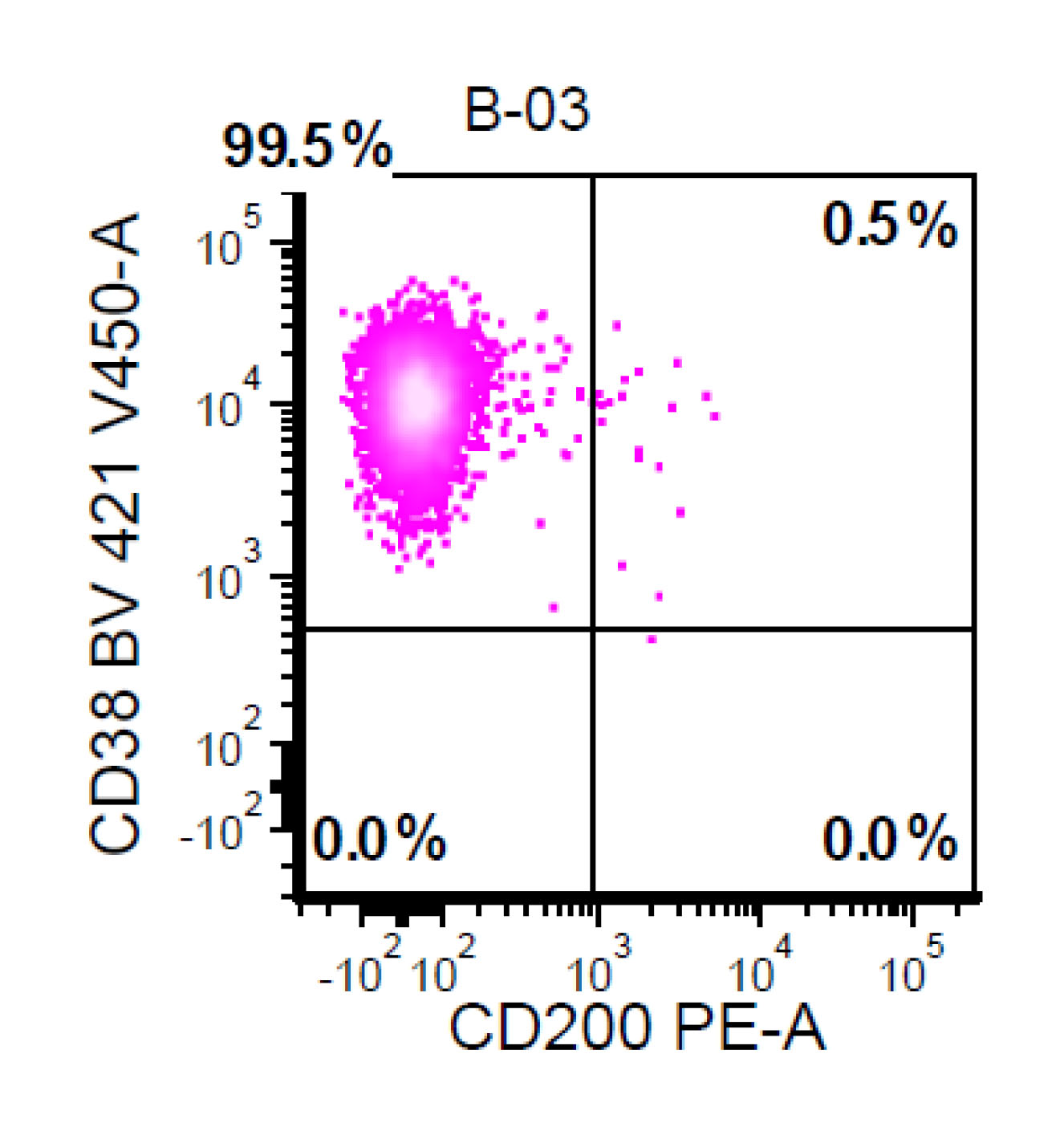
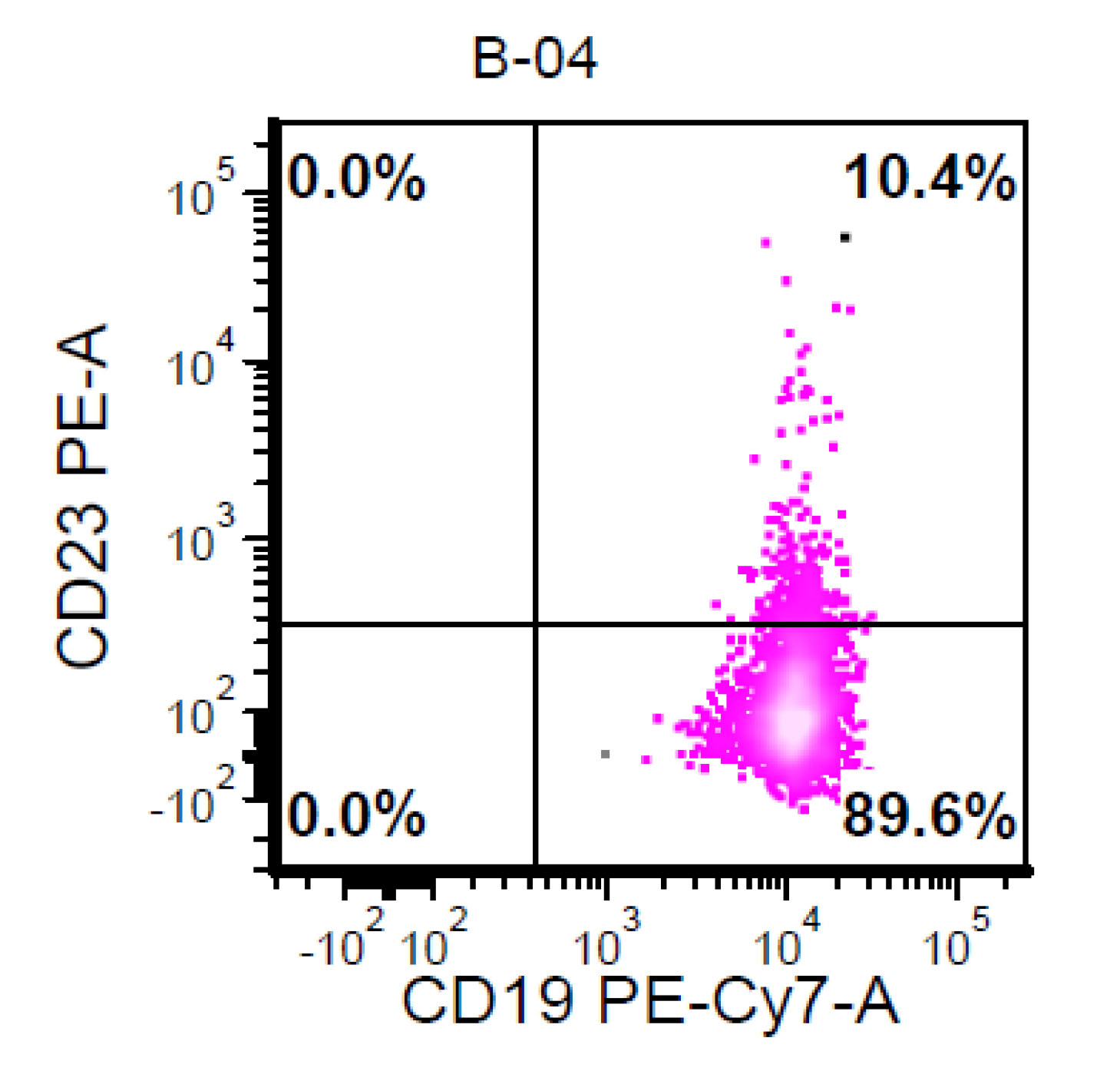
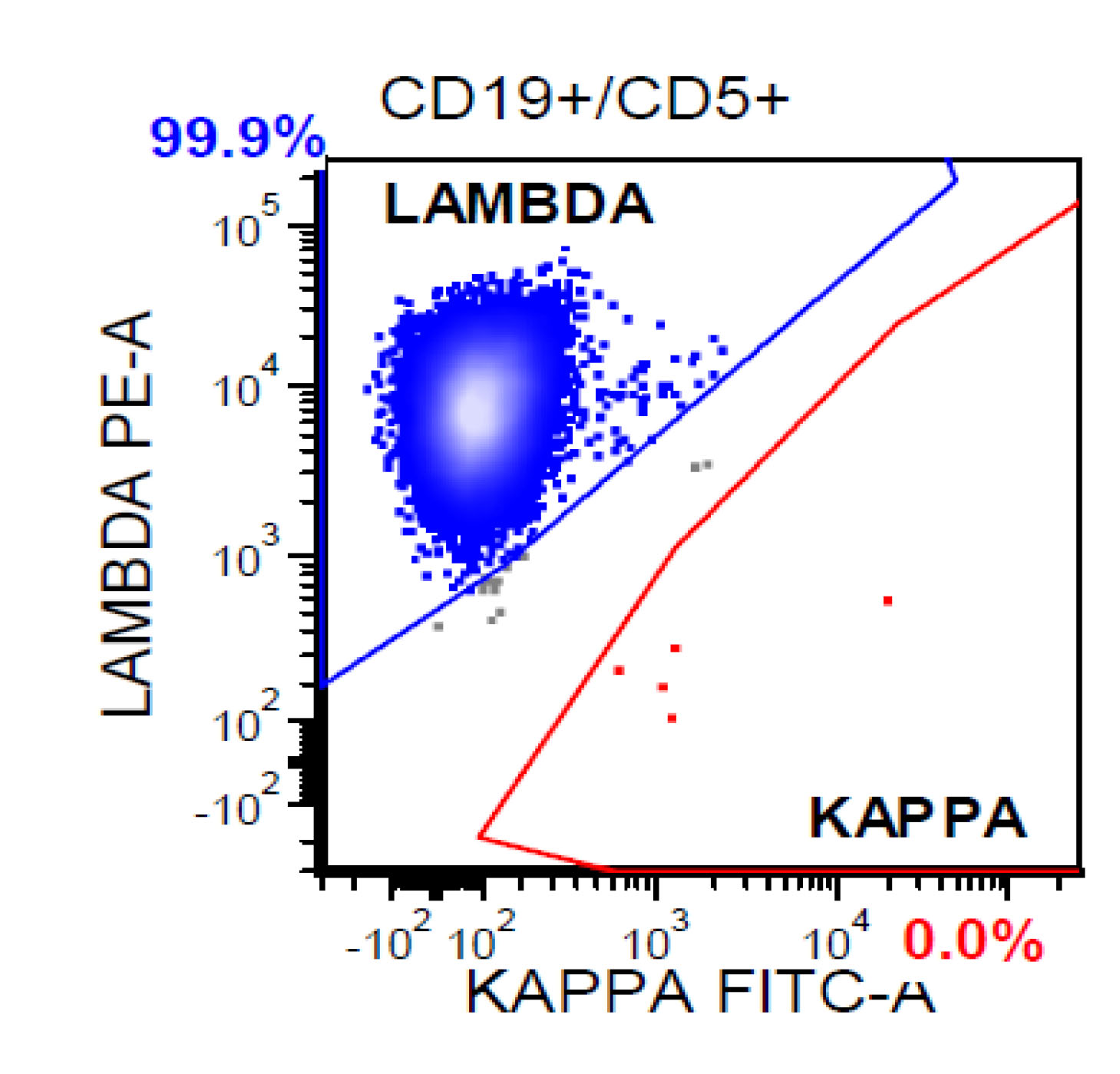
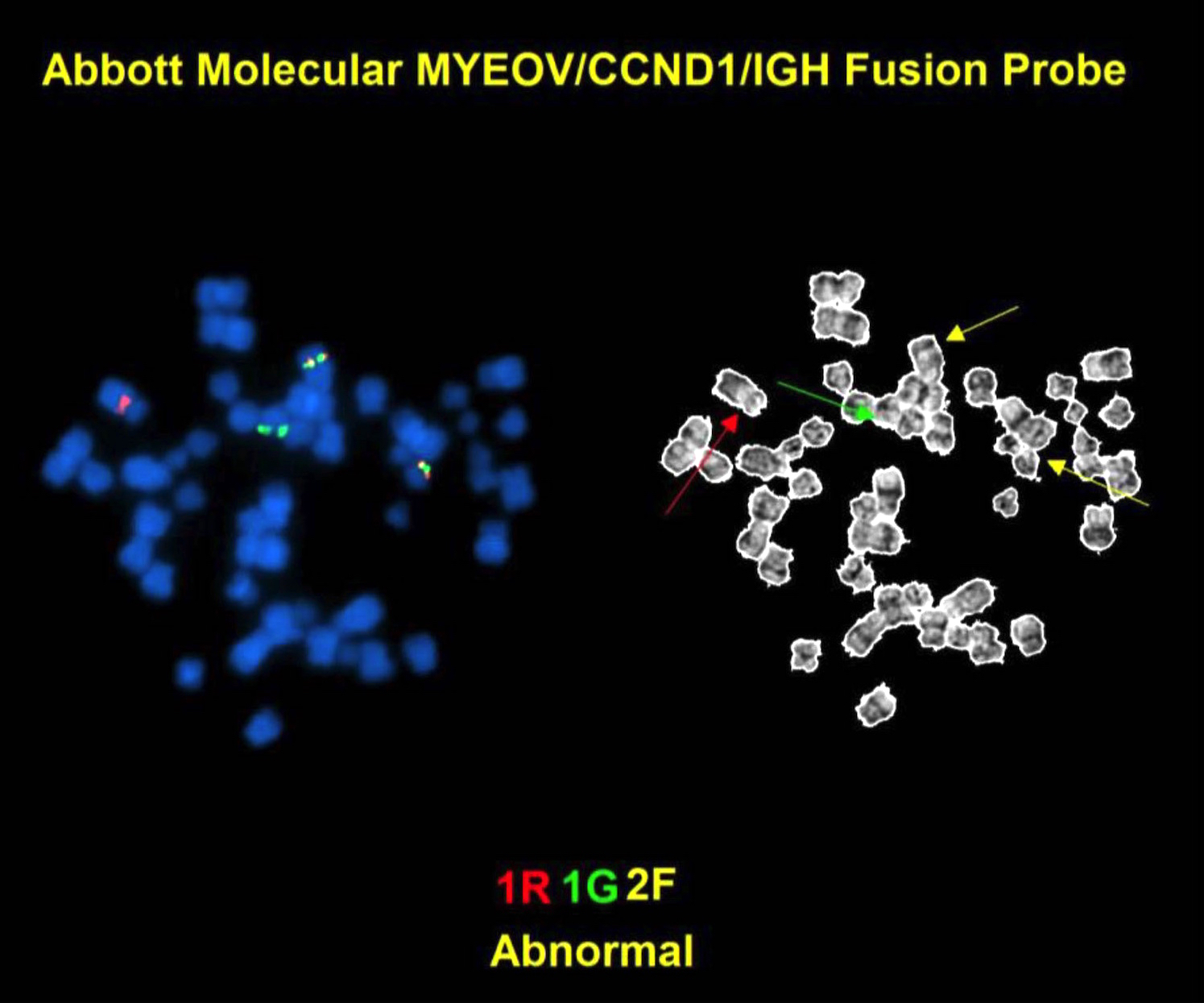
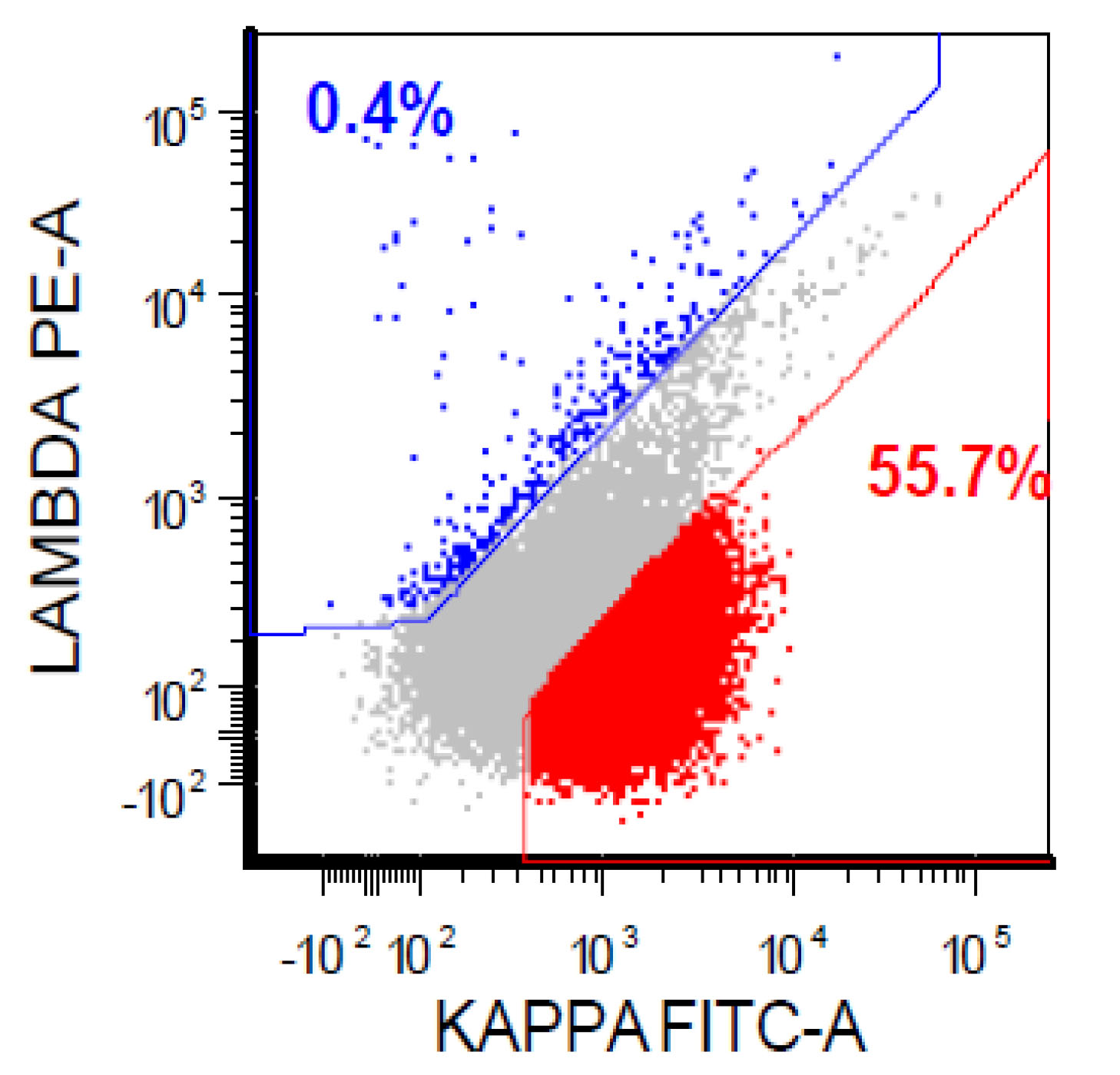
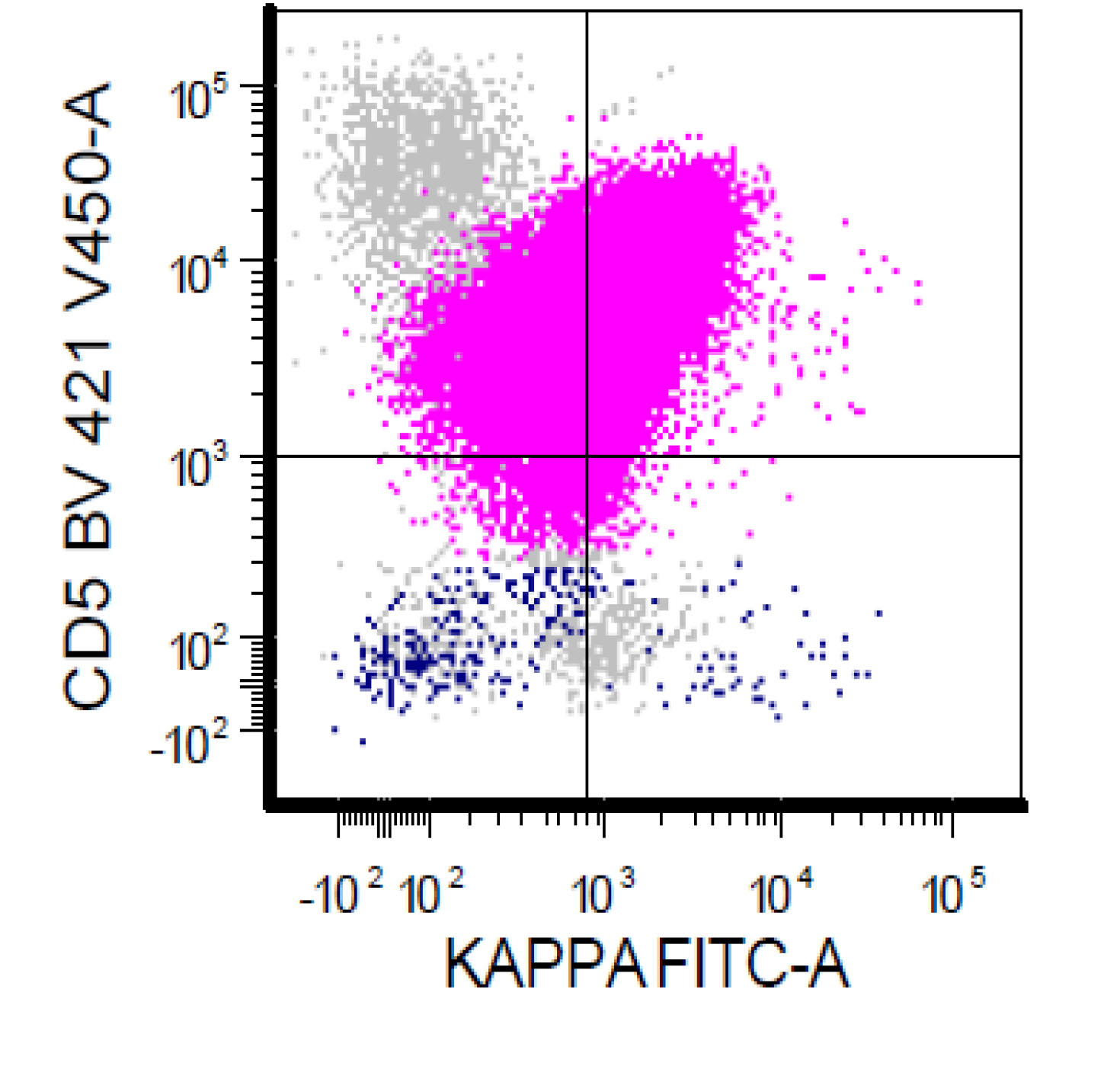
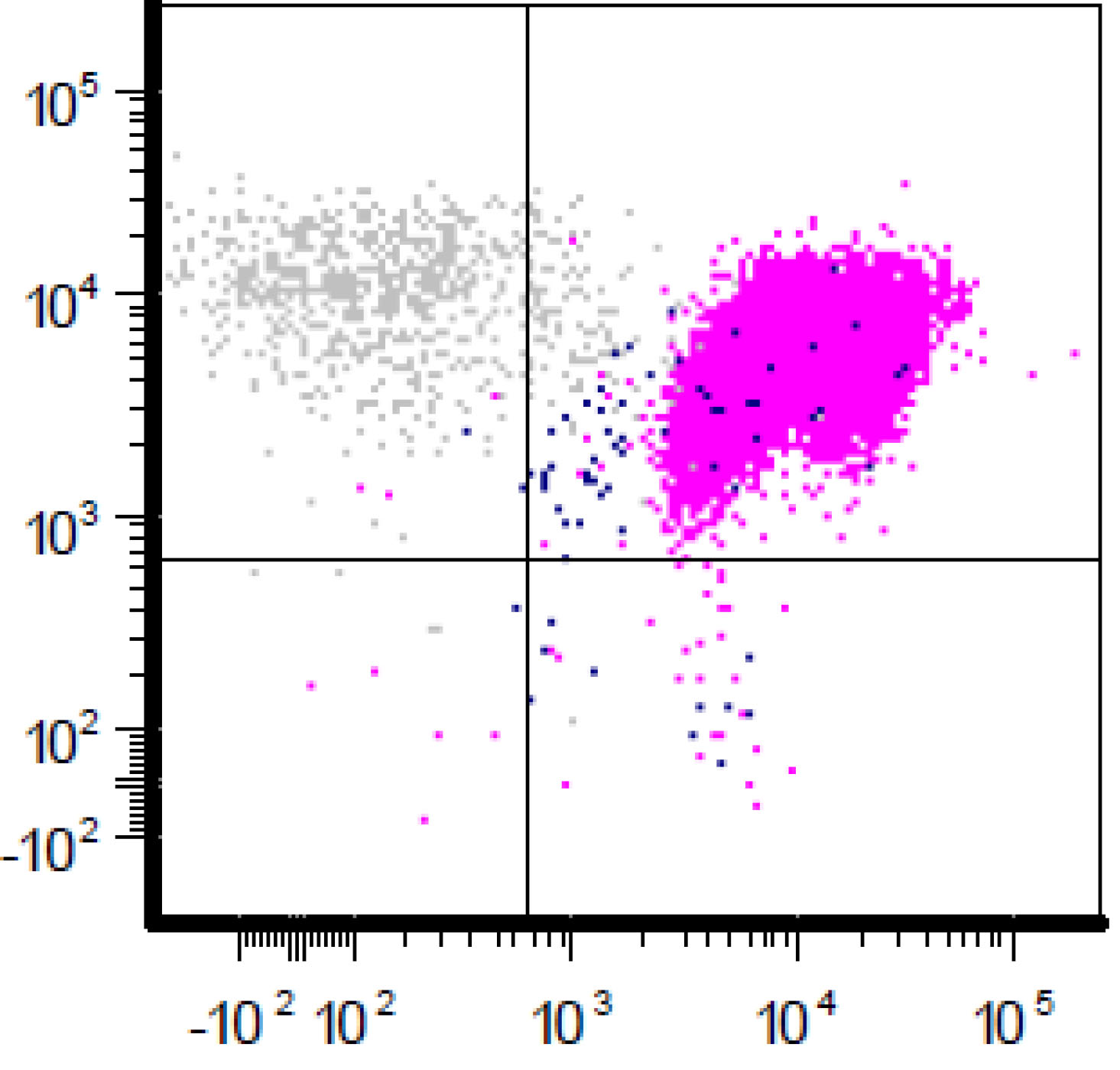
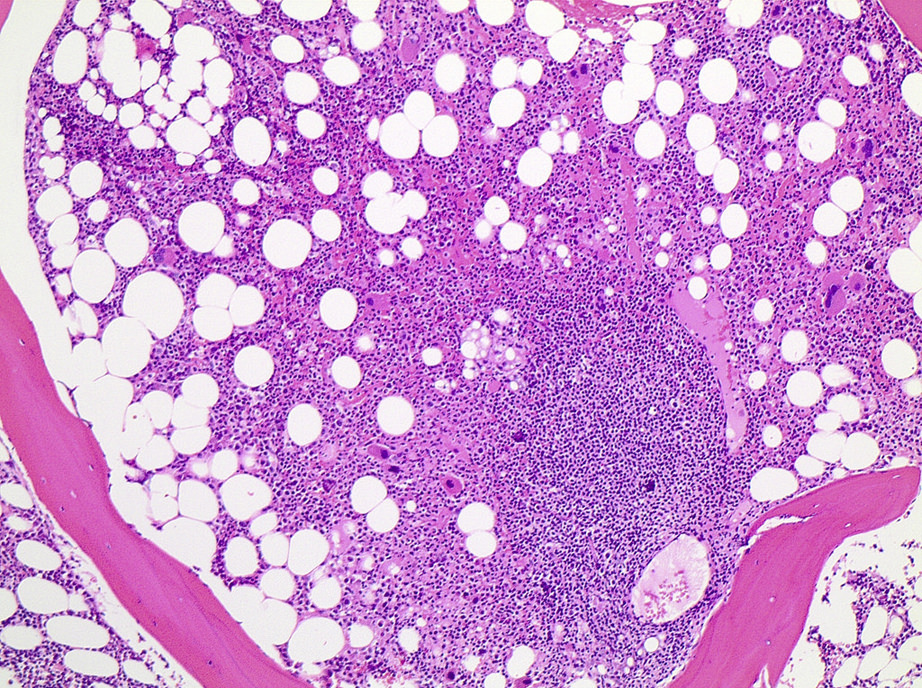
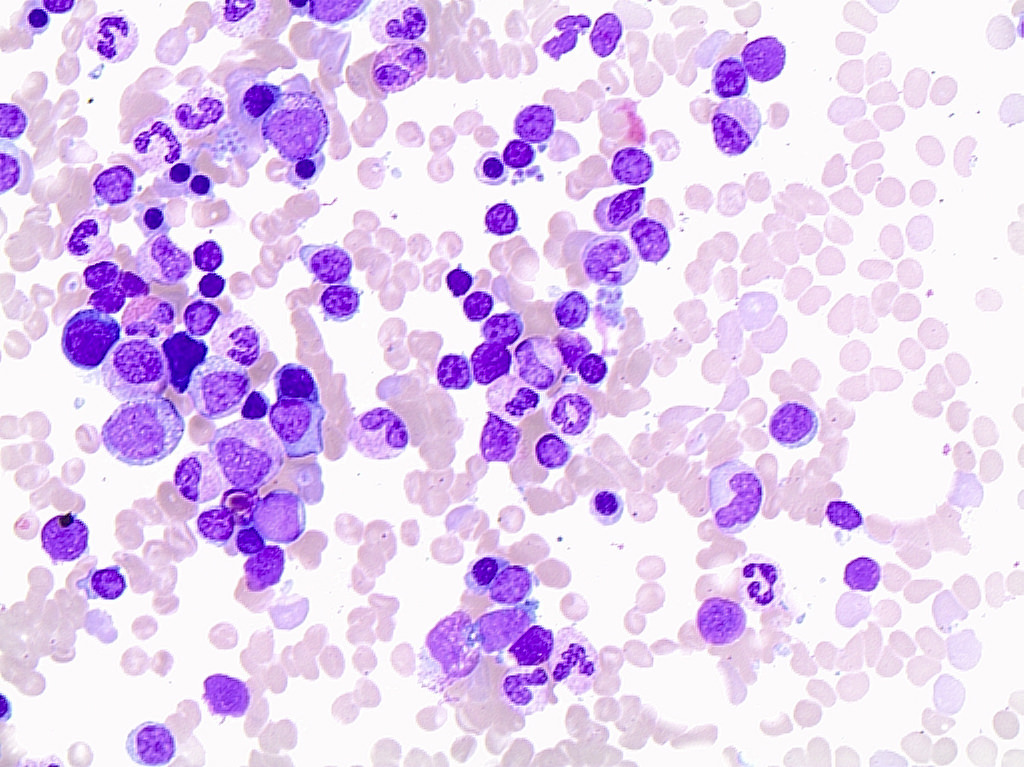
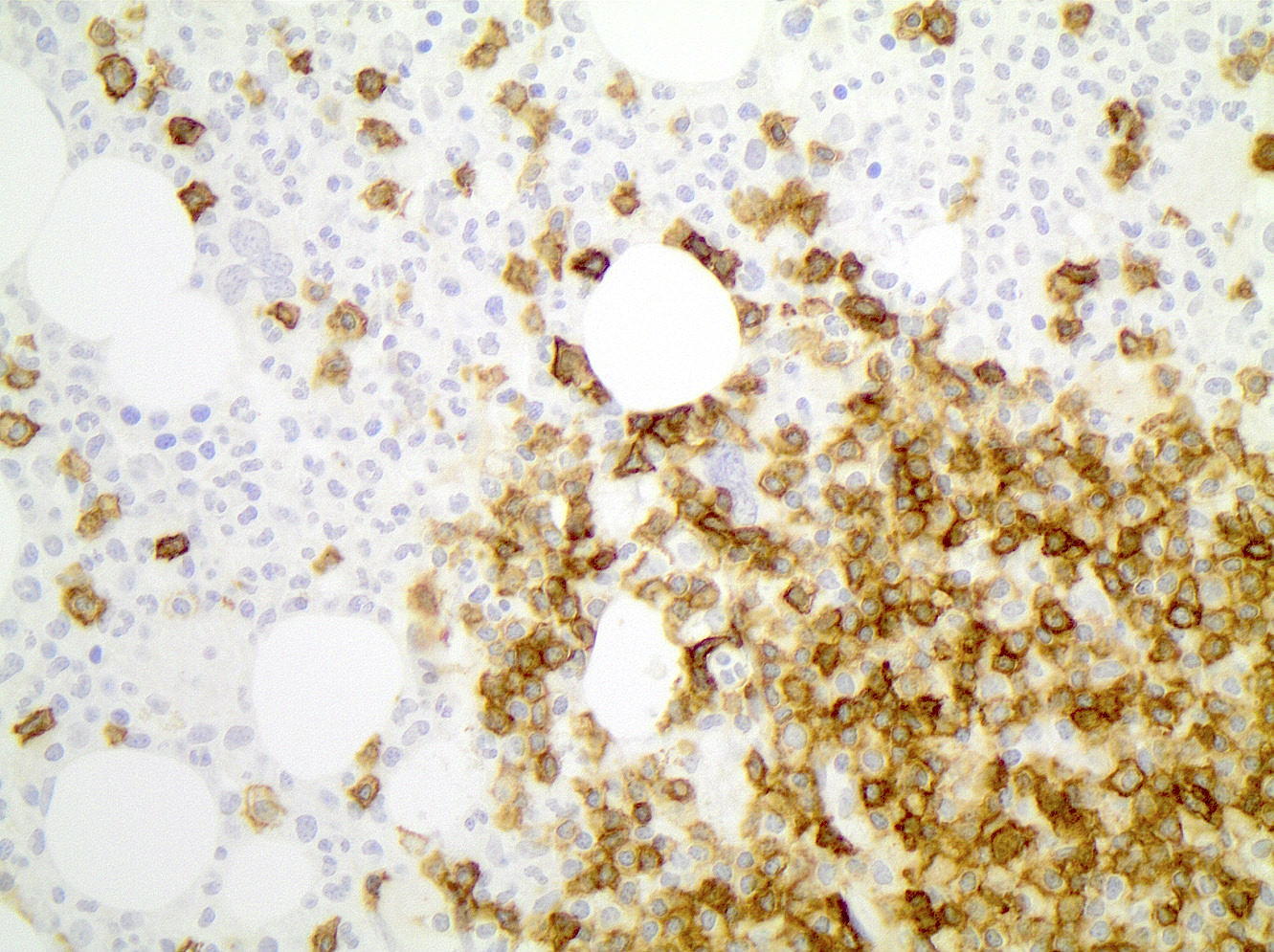
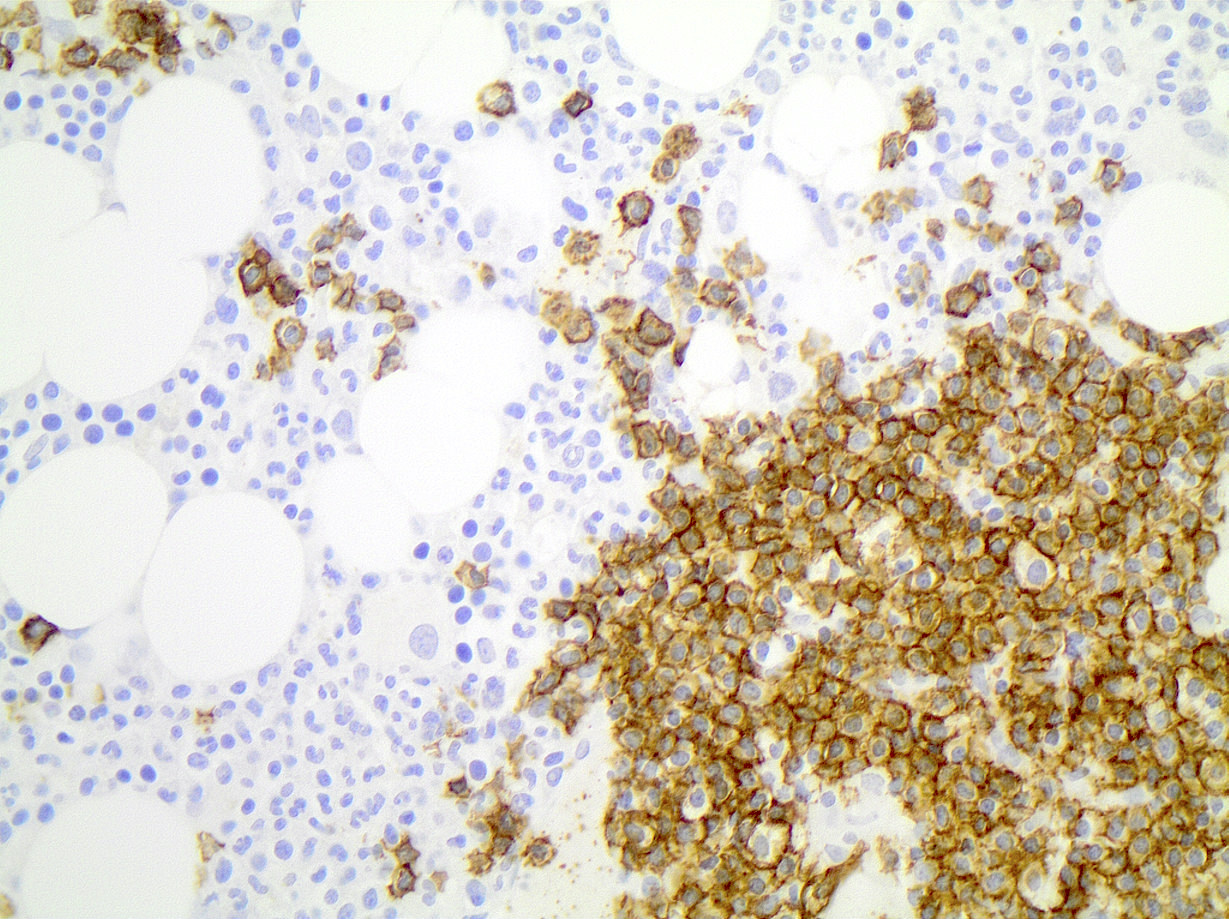
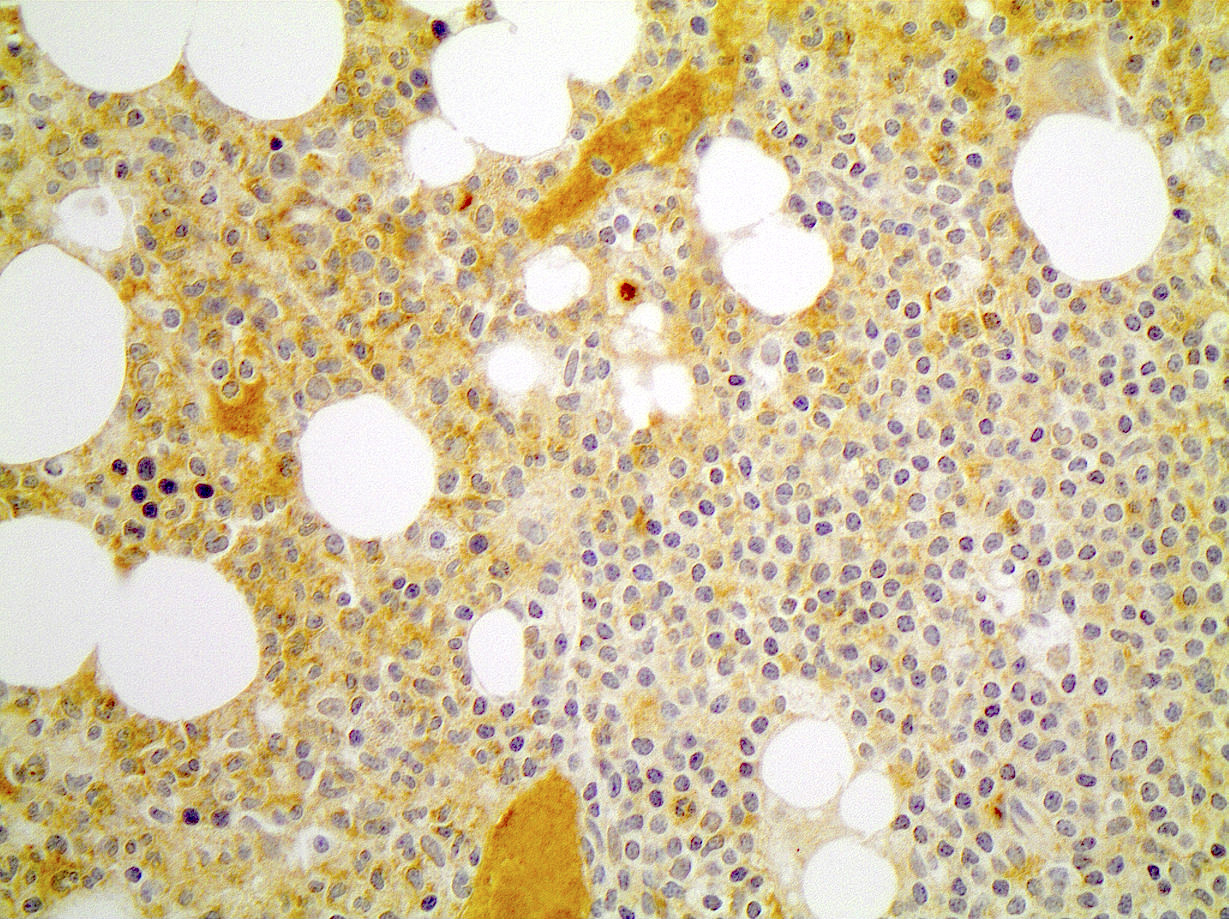
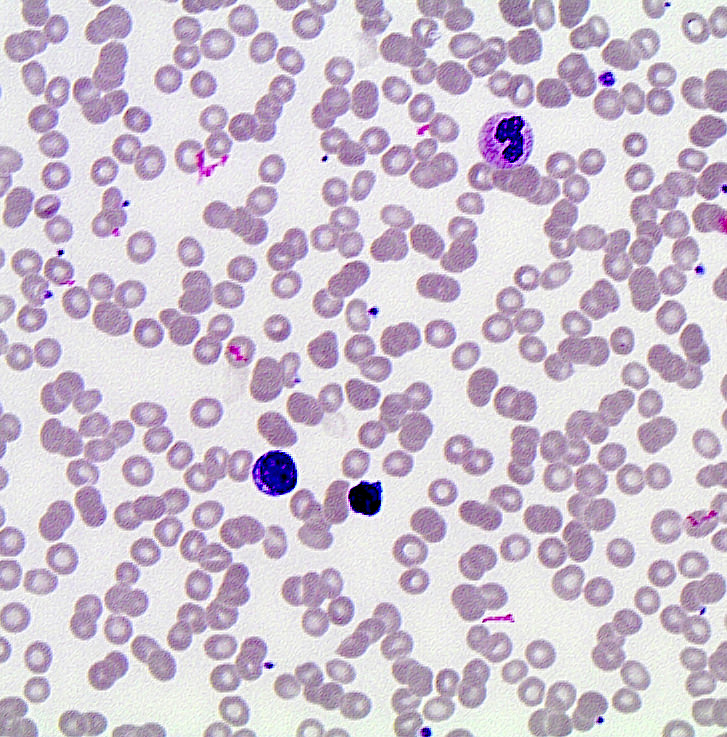
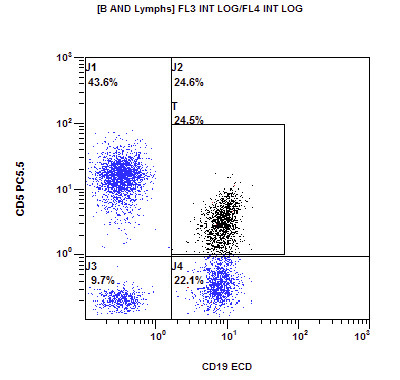
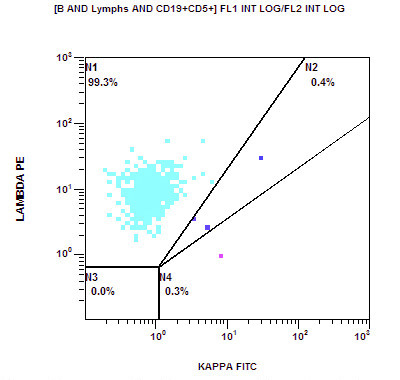
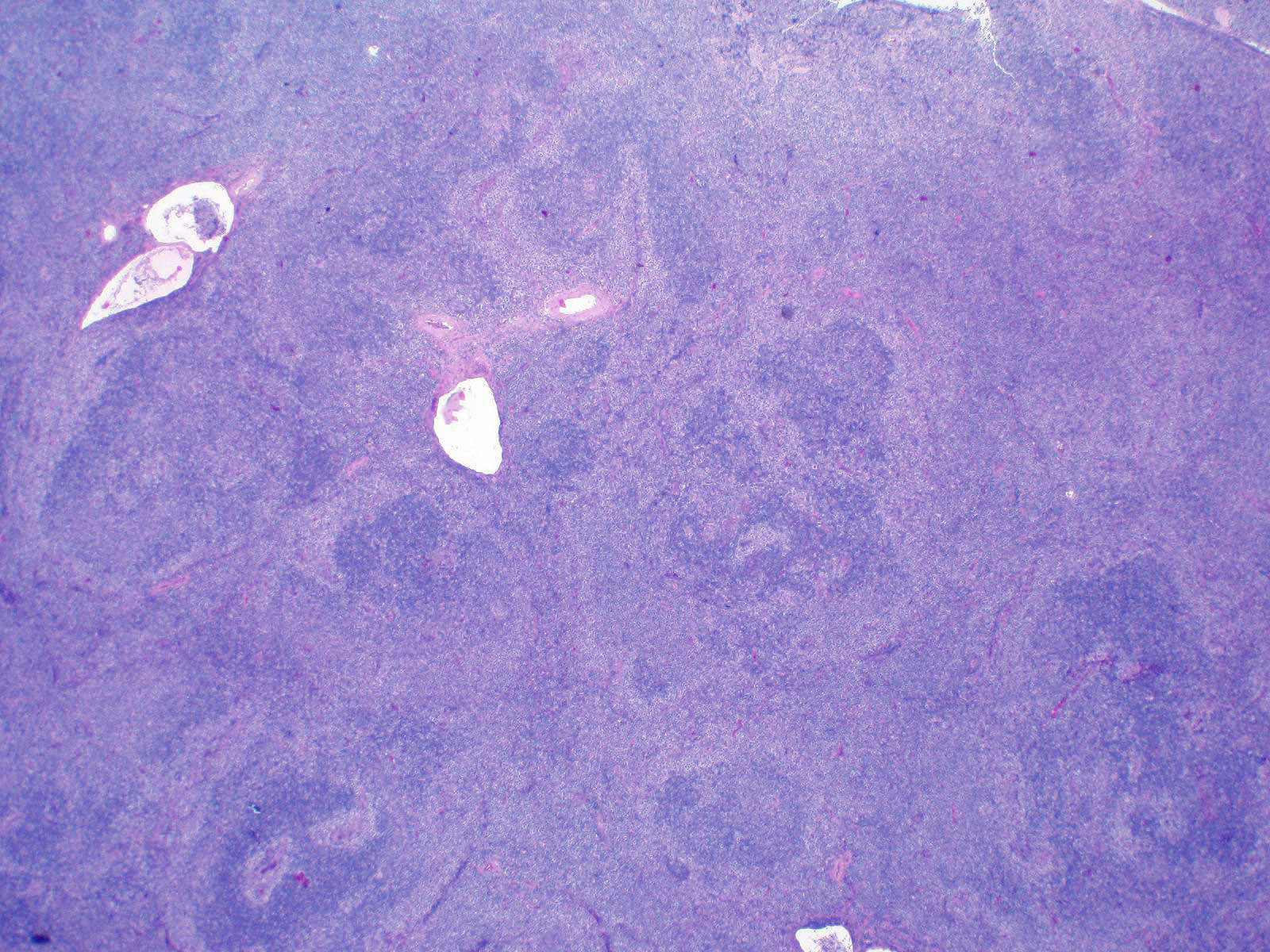
| Chronic lymphocytic leukemia / small lymphocytic lymphoma (CLL / SLL)
| Chronic lymphocytic leukemia / small lymphocytic lymphoma (CLL / SLL)
| Chronic lymphocytic leukemia / small lymphocytic lymphoma (CLL / SLL)
|
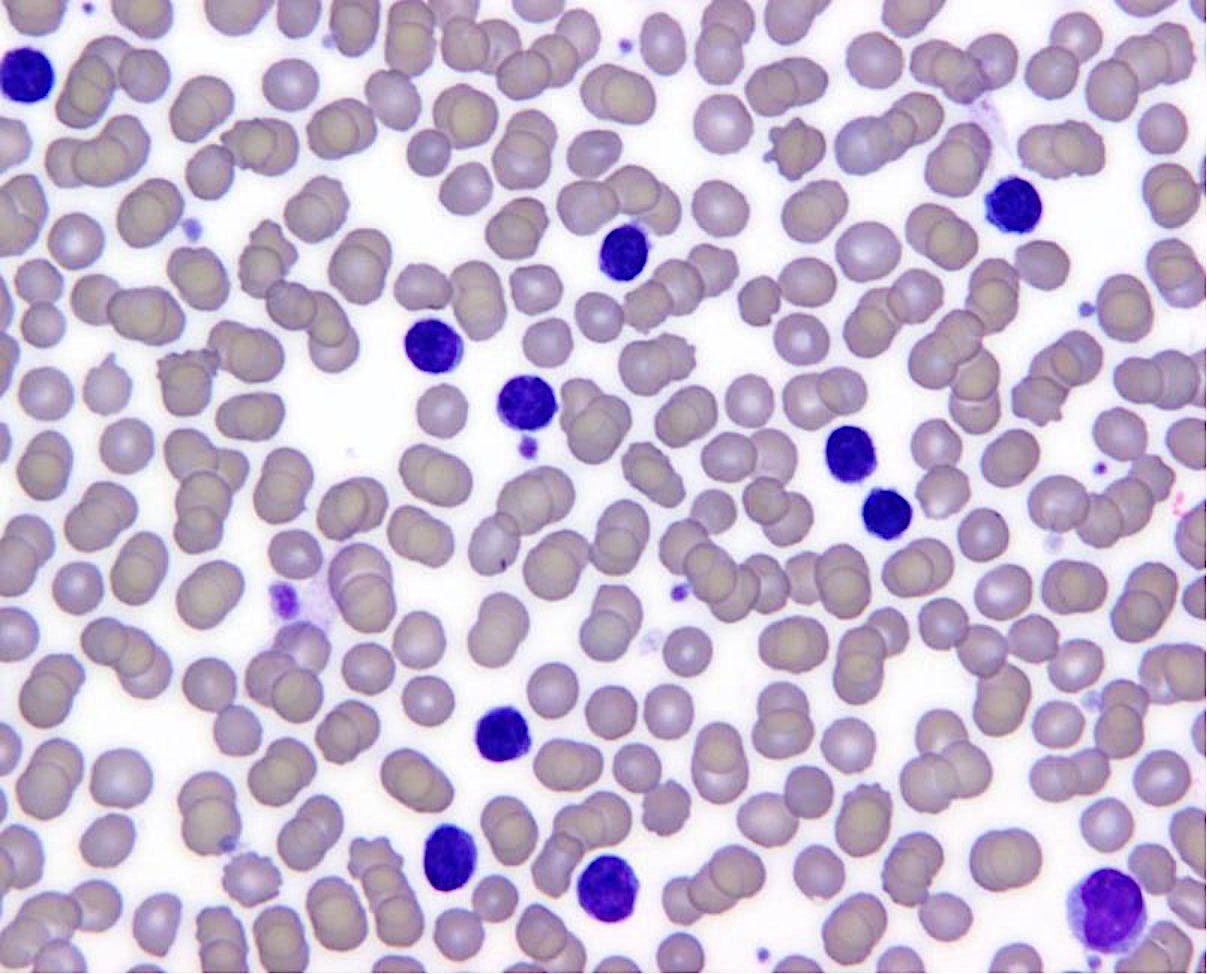
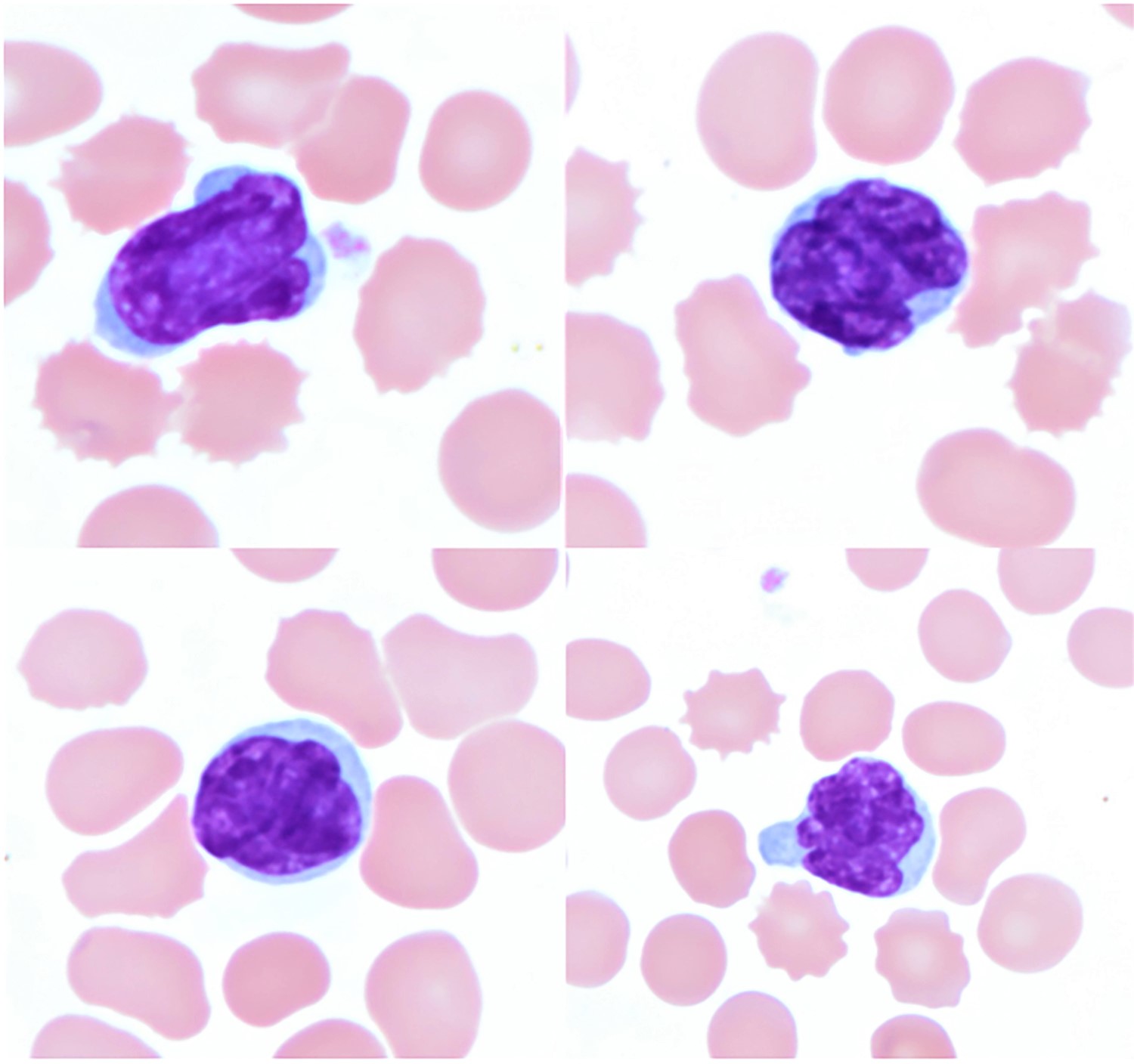
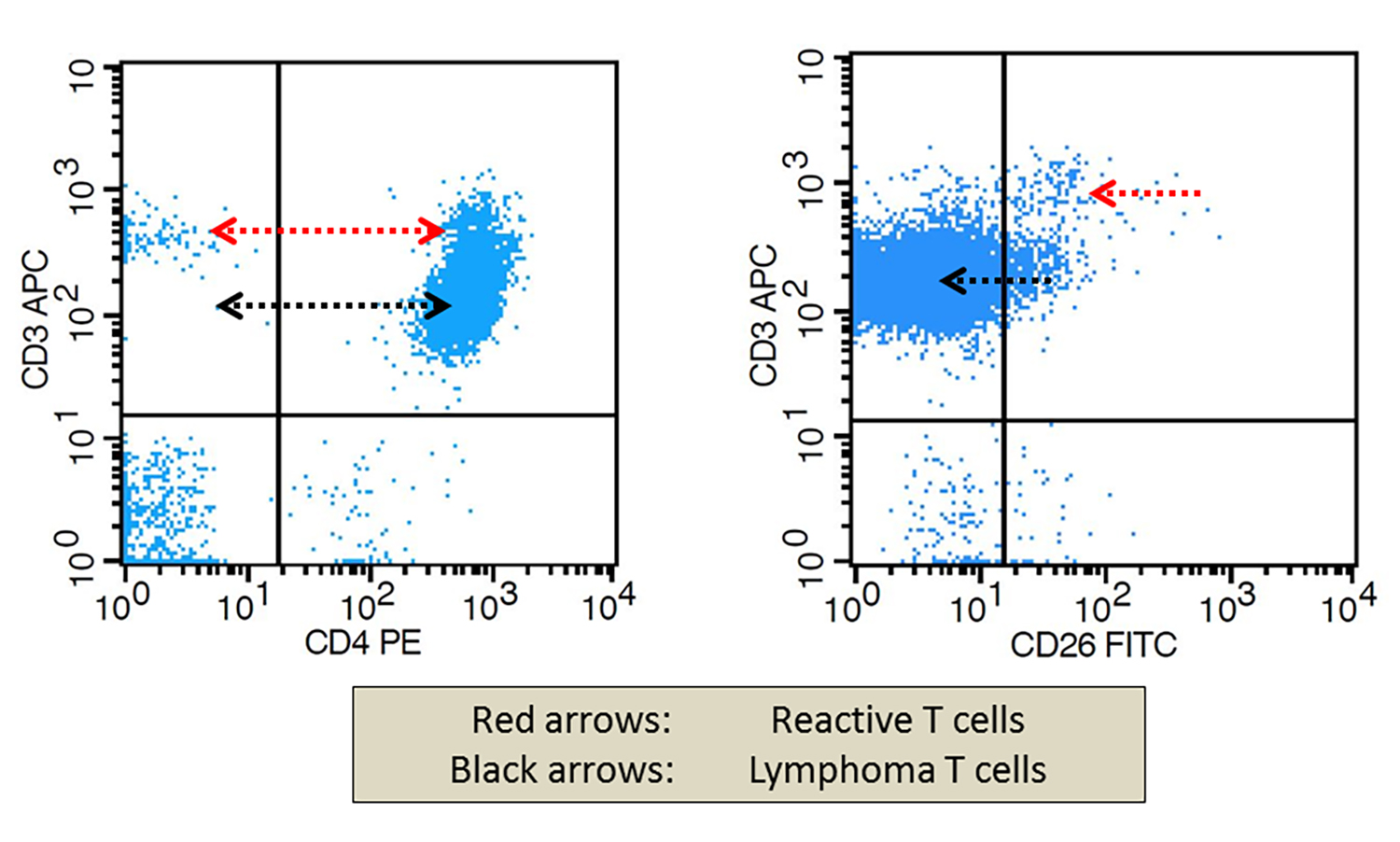
Monoclonal B cell lymphocytosis - CLL type
- Atypical CLL type
- Non-CLL type
| Monoclonal B cell lymphocytosis - Low count or clonal B cell expansion
- CLL / SLL type
- Non-CLL / SLL type
| Monoclonal B cell lymphocytosis
|
| B prolymphocytic leukemia (B PLL)
| Not a distinct entity but heterogeneous
| B prolymphocytic leukemia (B PLL)
|
Lymphoplasmacytic lymphoma- Waldenström macroglobulinemia
| Lymphoplasmacytic lymphoma - Waldenström macroglobulinemia
- Non-Waldenström macroglobulinemia
| Lymphoplasmacytic lymphoma - Waldenström macroglobulinemia
|
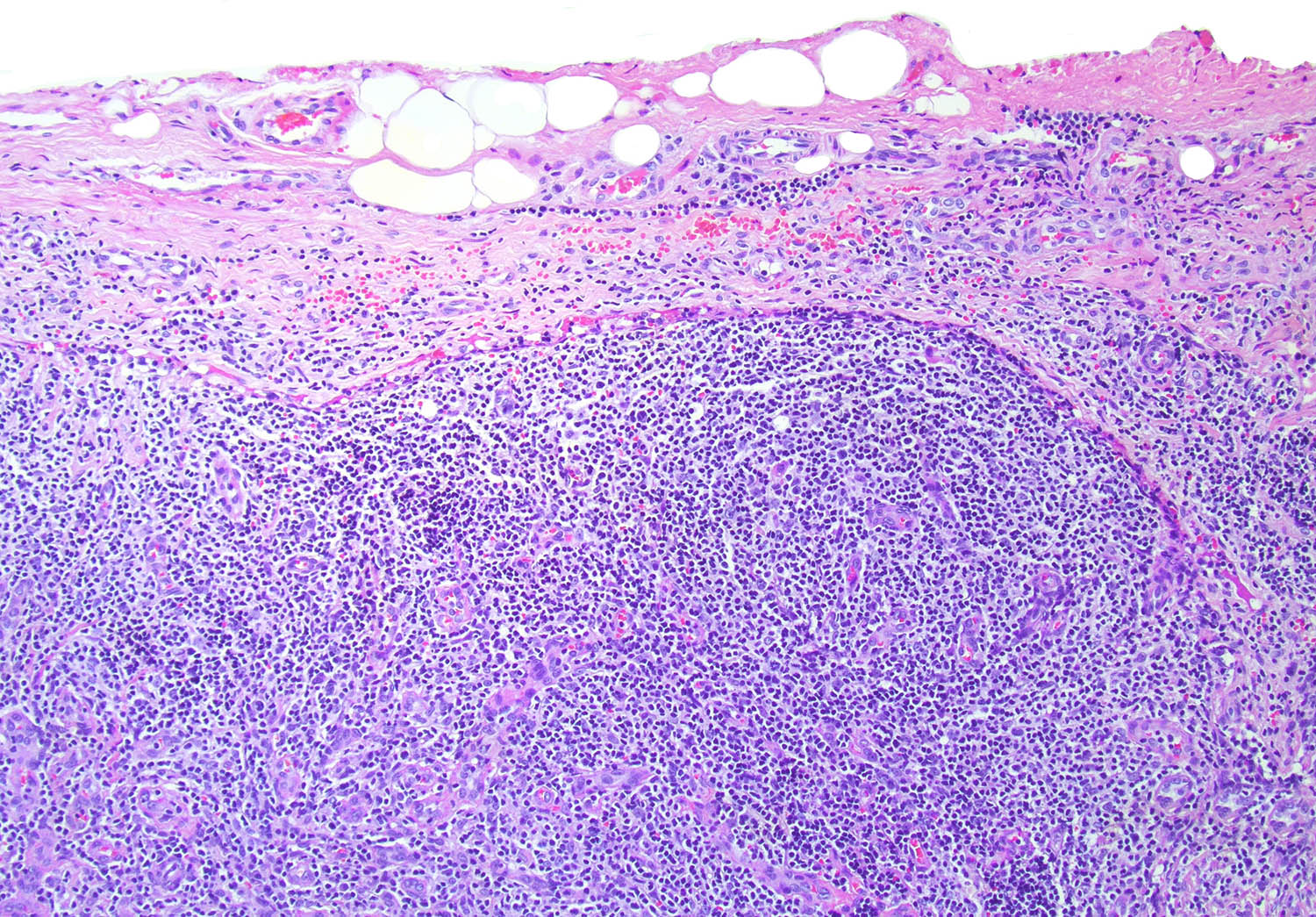
| Marginal zone lymphoma
|
| Nodal marginal zone lymphoma
| Nodal marginal zone lymphoma
| Nodal marginal zone lymphoma
|
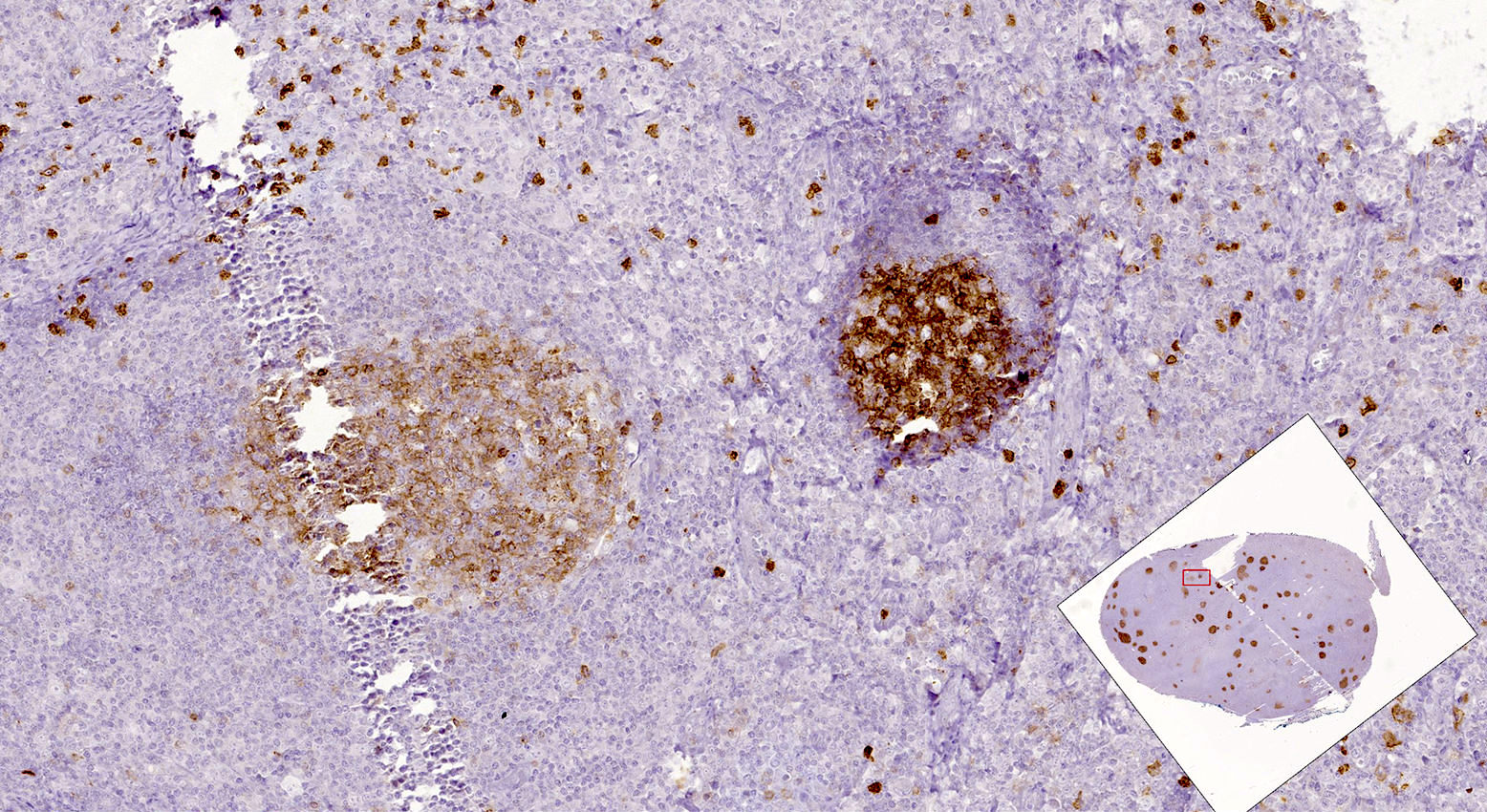
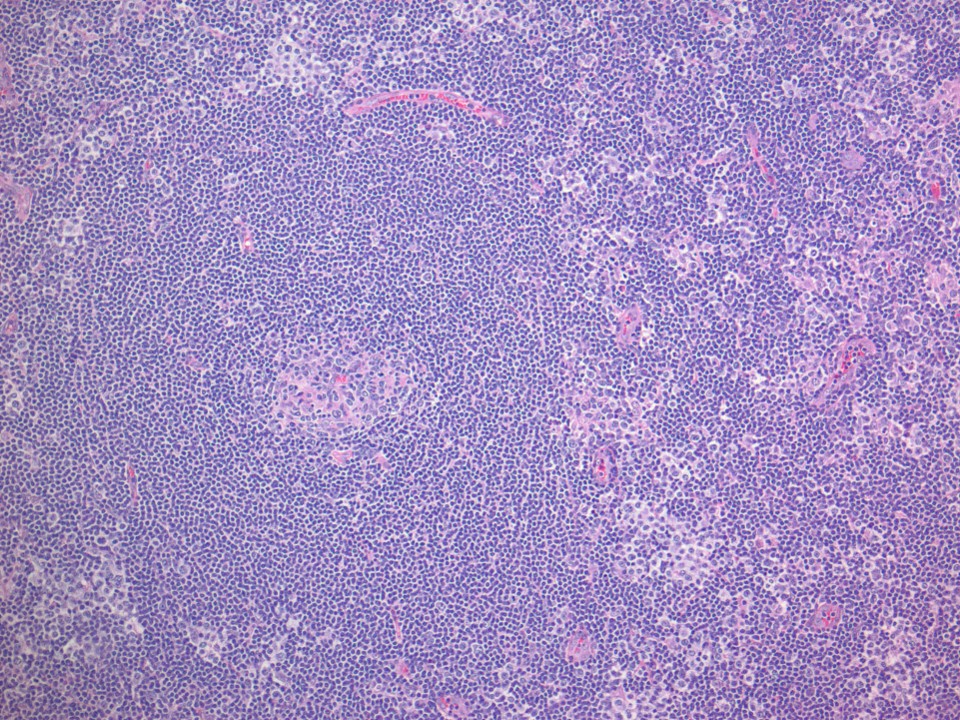
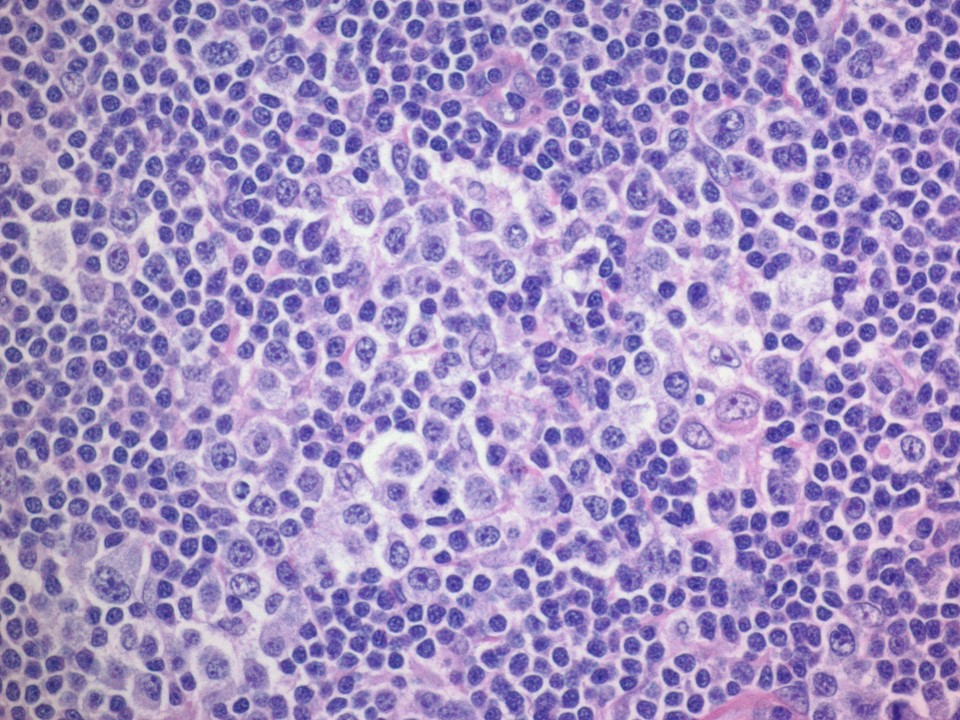
| Pediatric nodal marginal zone lymphoma*
| Pediatric nodal marginal zone lymphoma
| Pediatric nodal marginal zone lymphoma*
|
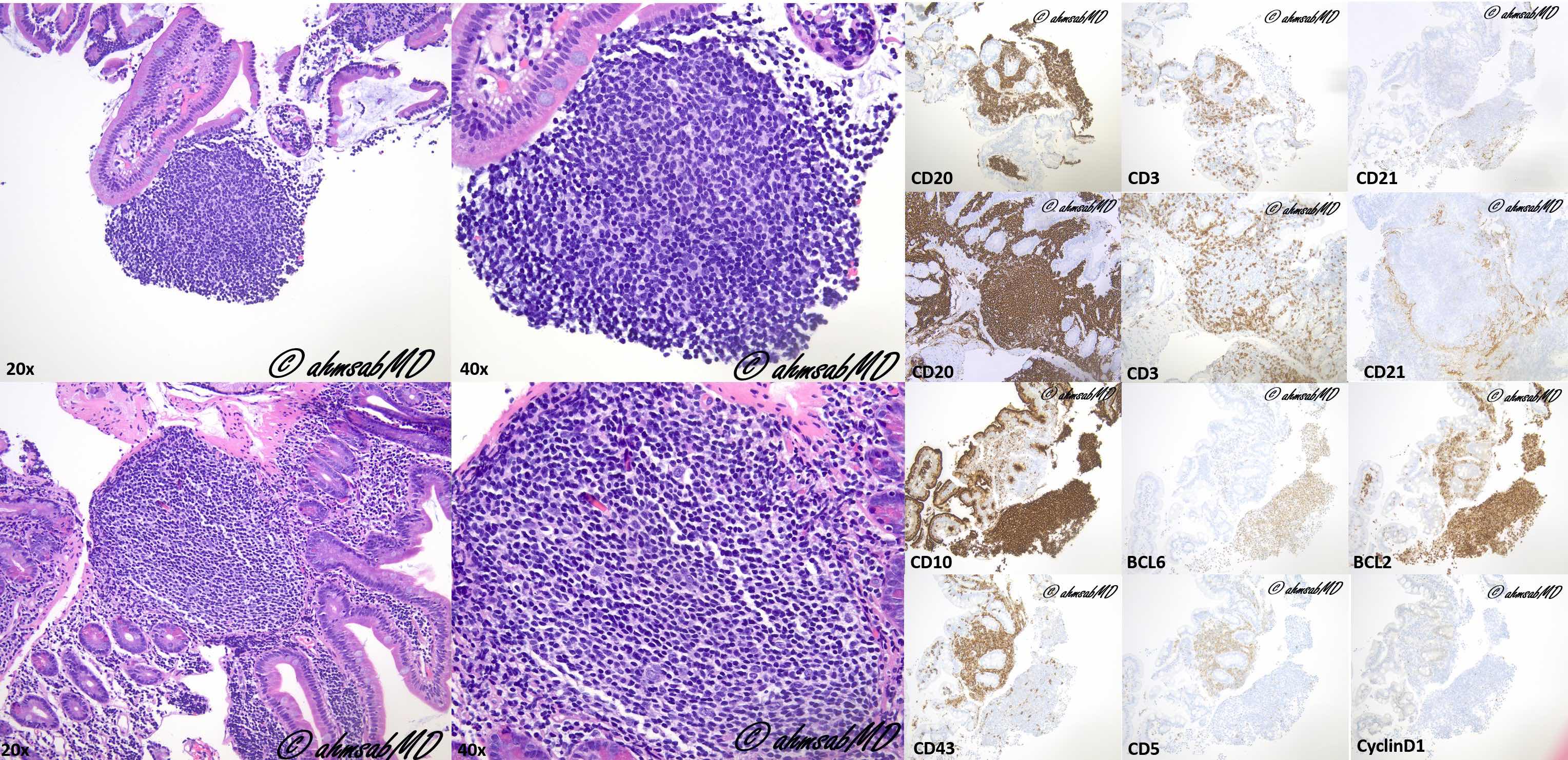
| Extranodal marginal zone lymphoma of mucosa associated lymphoid tissue (MALT)
| Extranodal marginal zone lymphoma of mucosa associated lymphoid tissue (MALT)
| Extranodal marginal zone lymphoma of mucosa associated lymphoid tissue (MALT)
|
| Not included; part of extranodal marginal zone lymphoma of MALT
| Primary cutaneous marginal zone lymphoma
| Primary cutaneous marginal zone lymphoproliferative disorder
|
| Splenic B cell lymphoma / leukemia
|
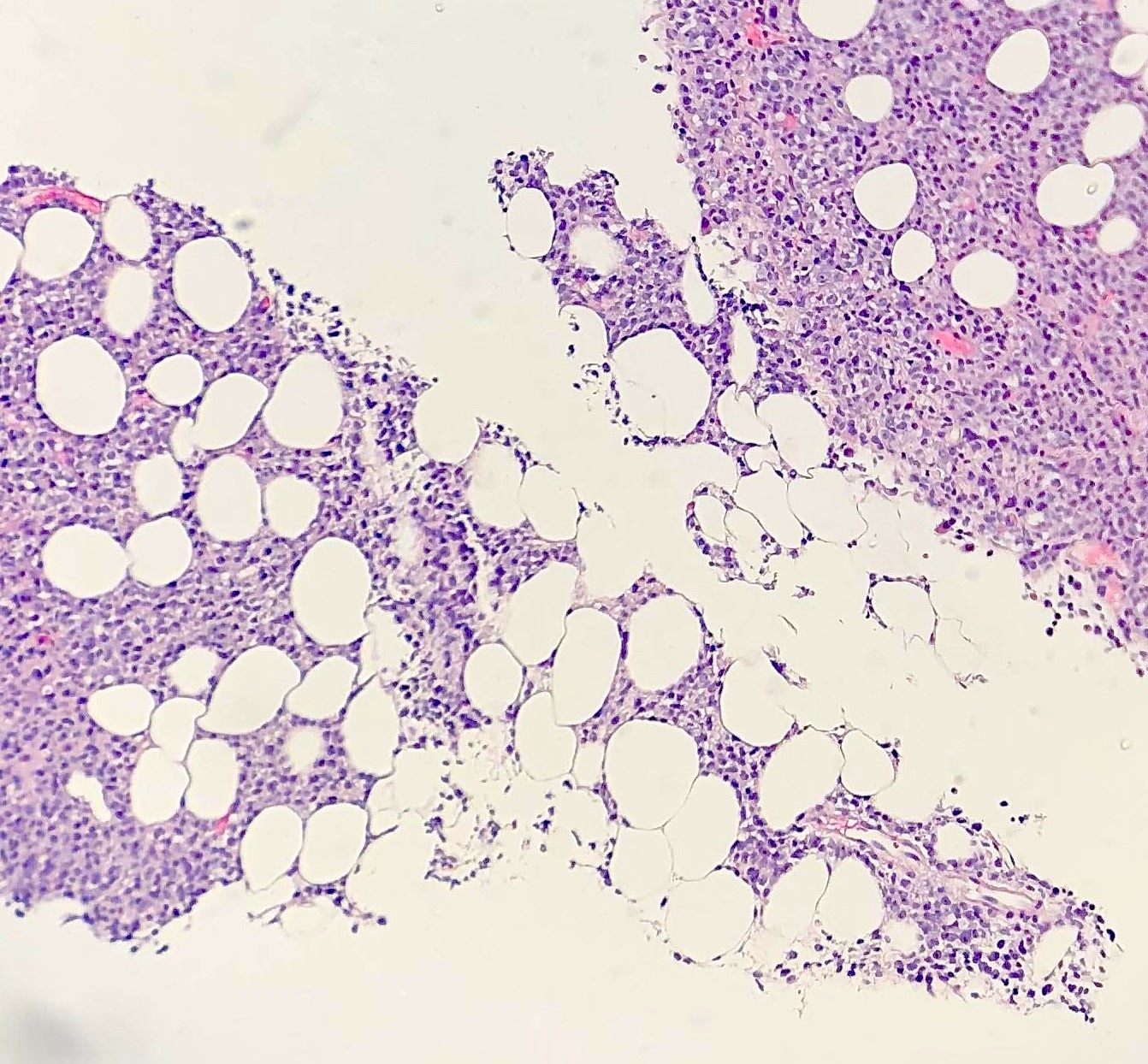
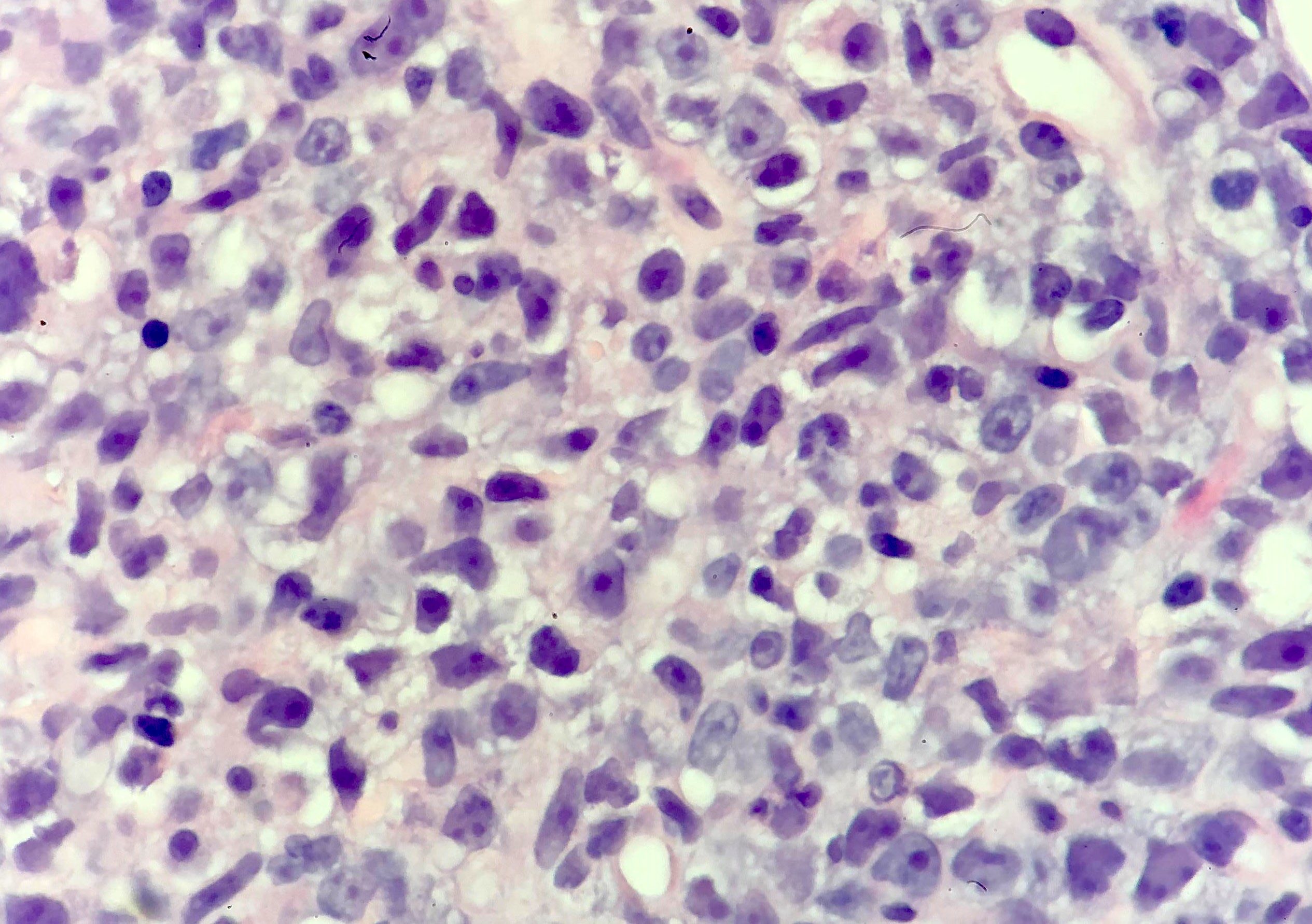
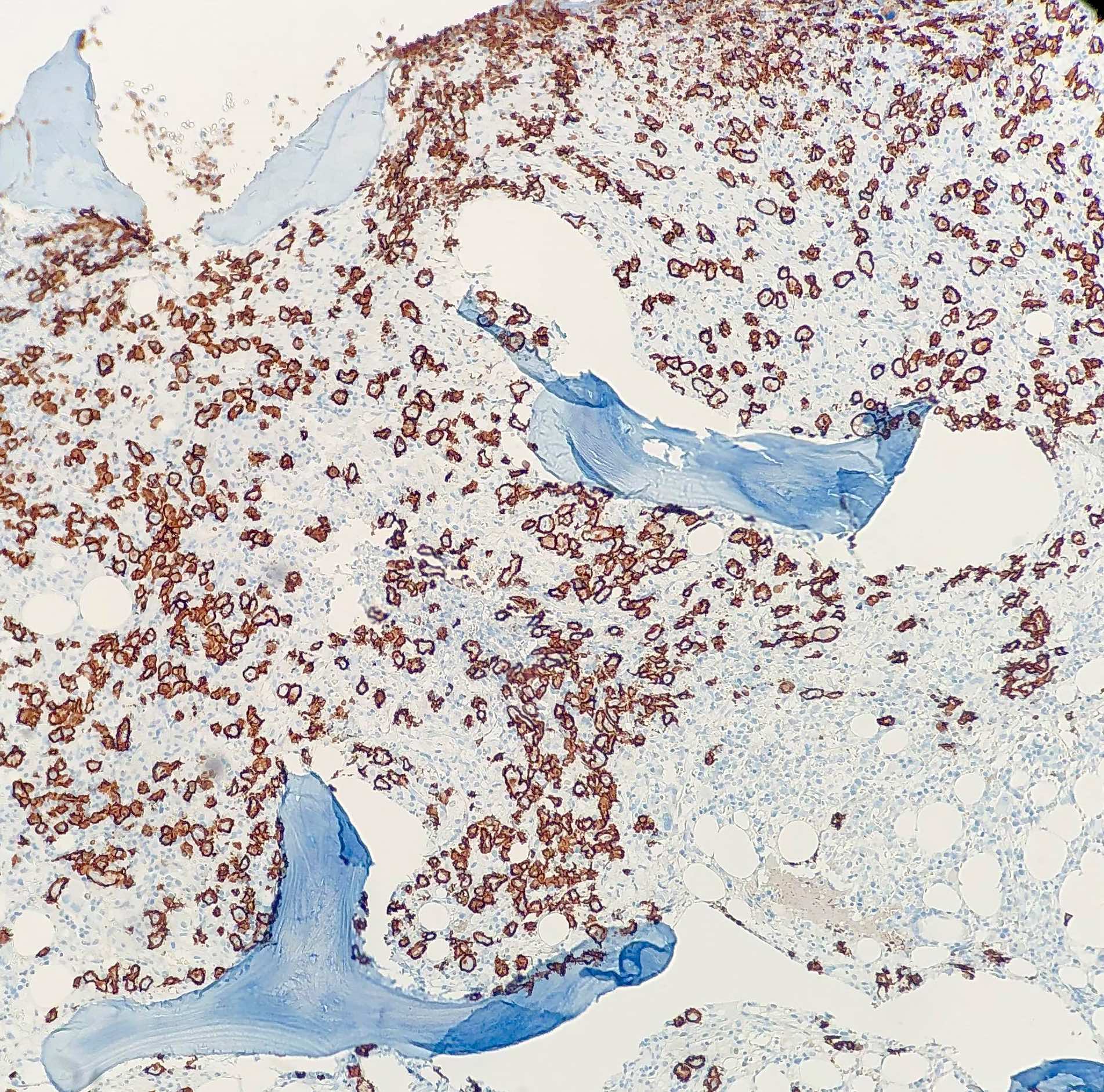
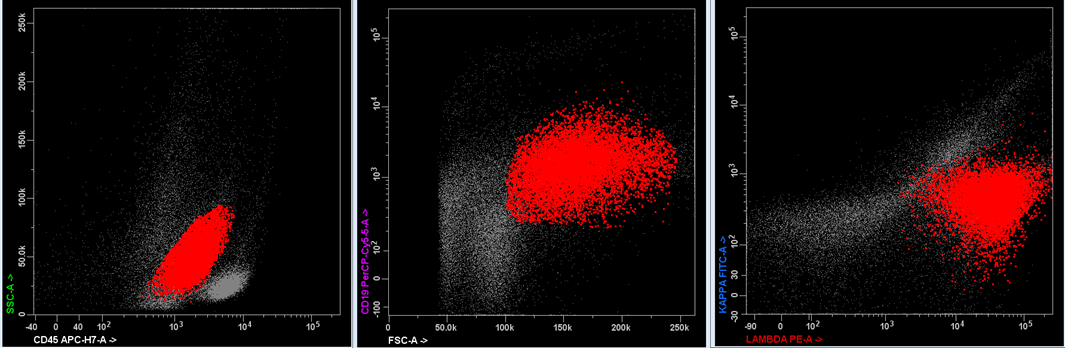
| Splenic marginal zone lymphoma
| Splenic marginal zone lymphoma
| Splenic marginal zone lymphoma
|
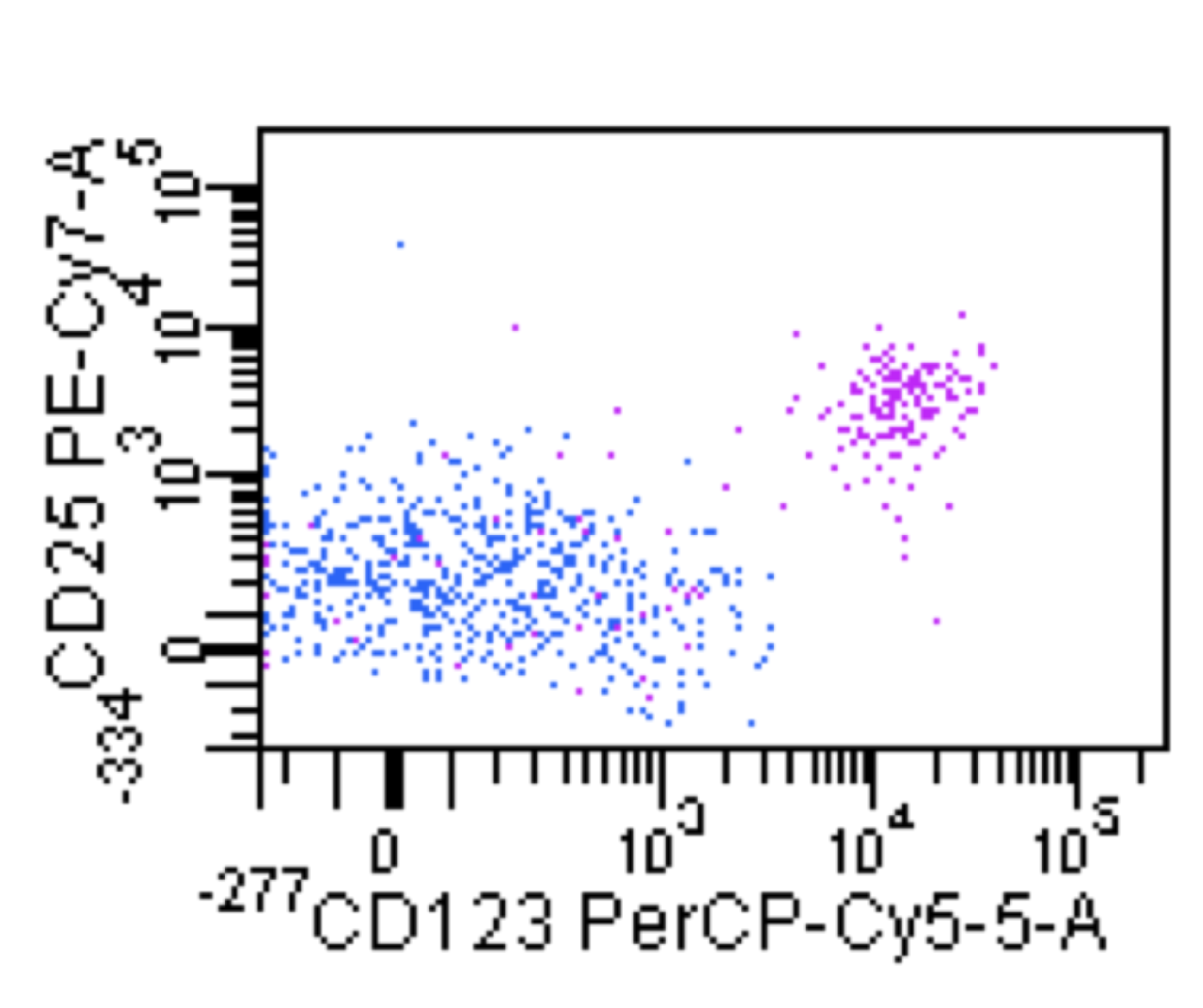
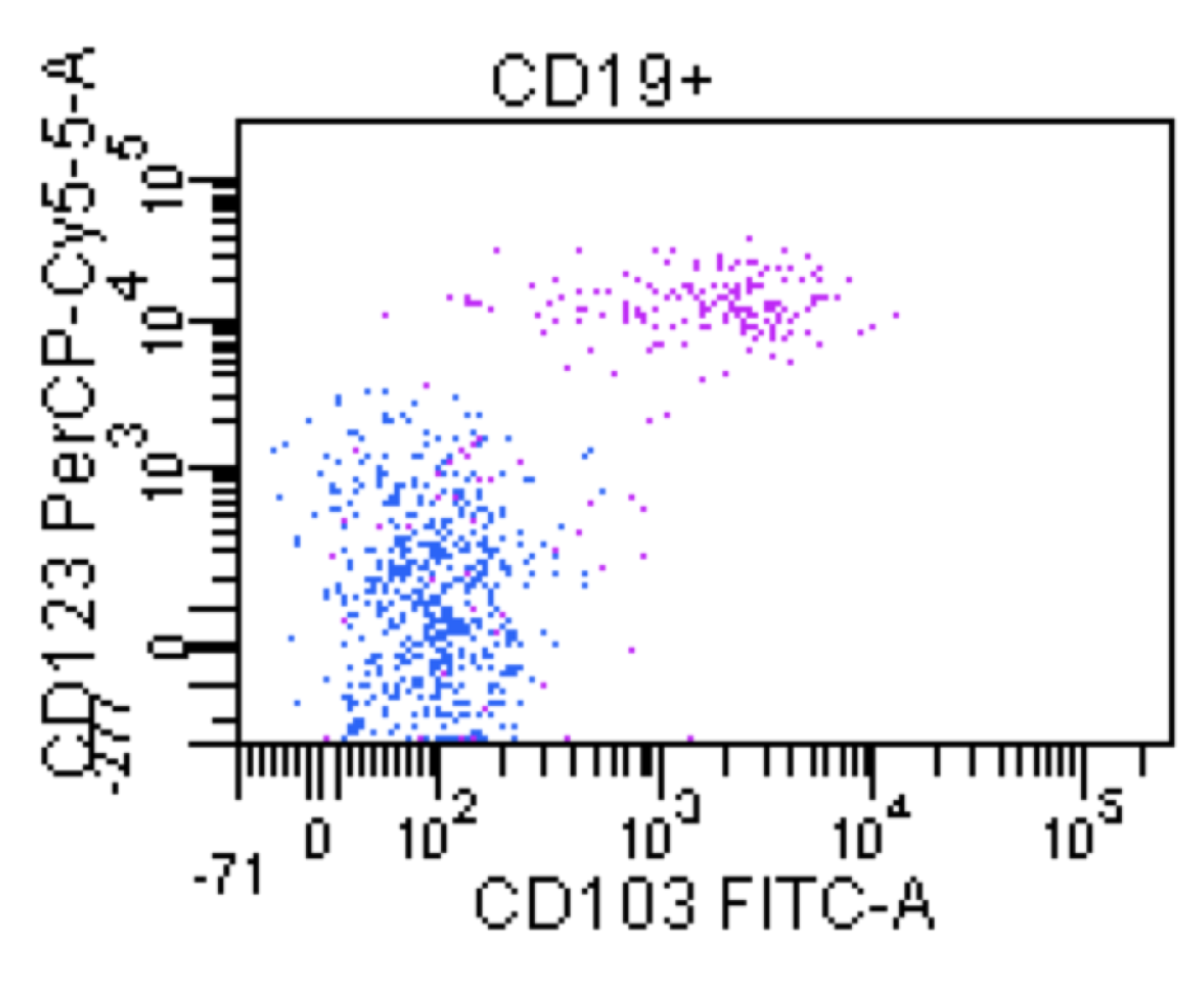
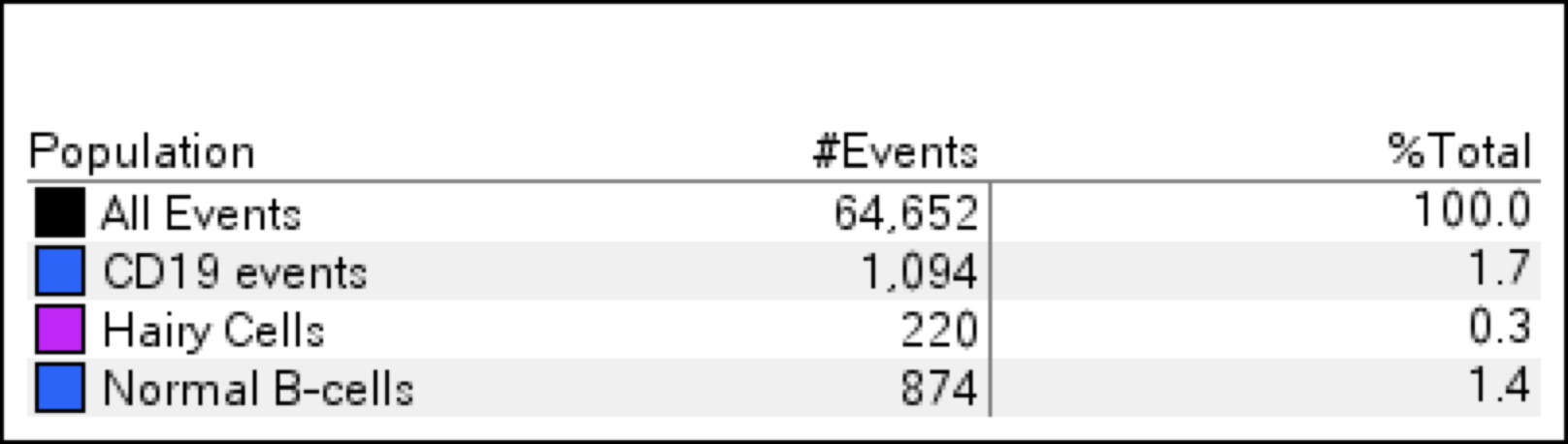
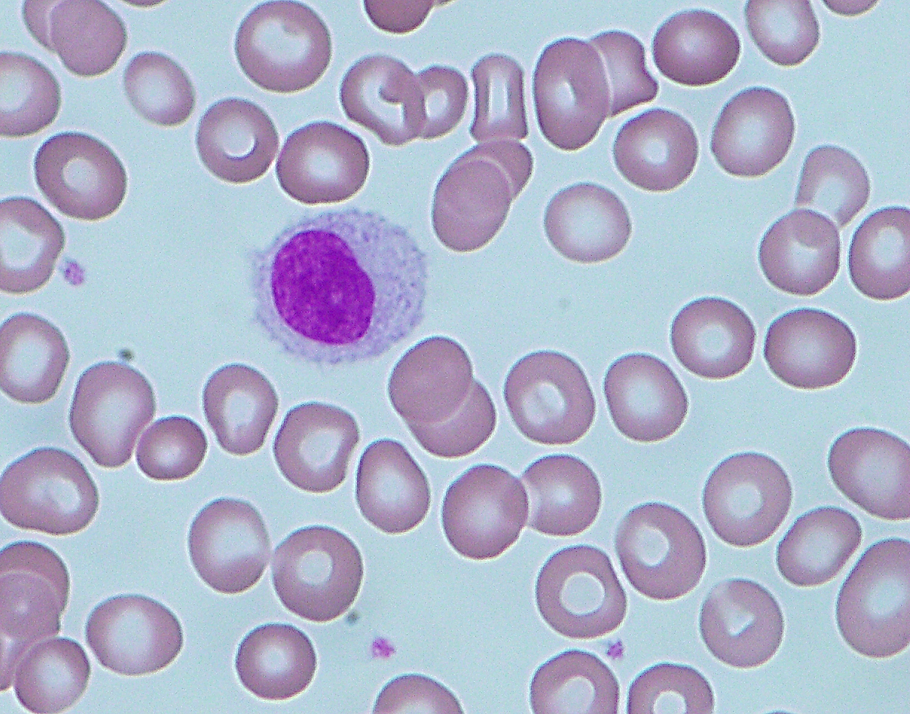
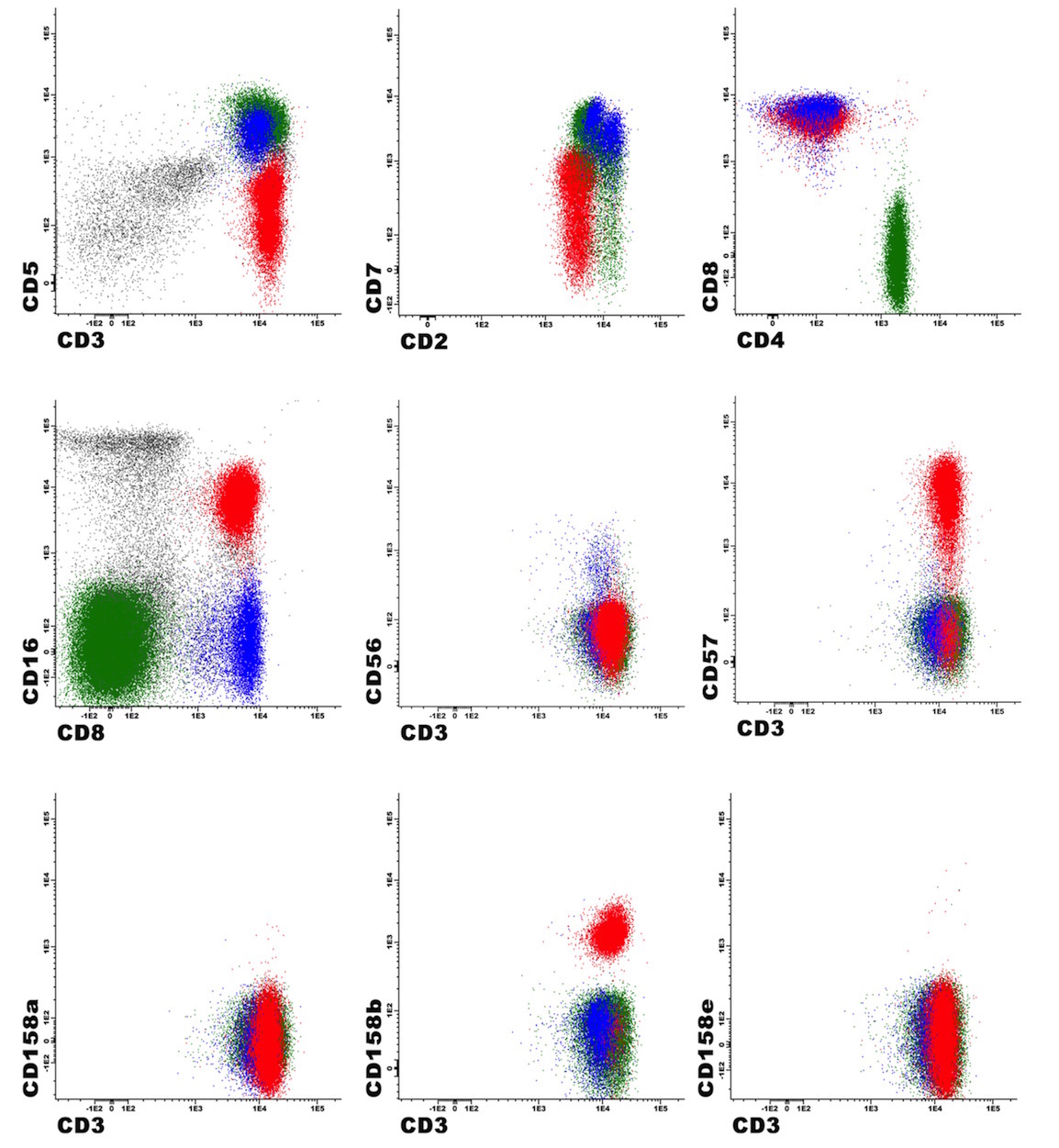
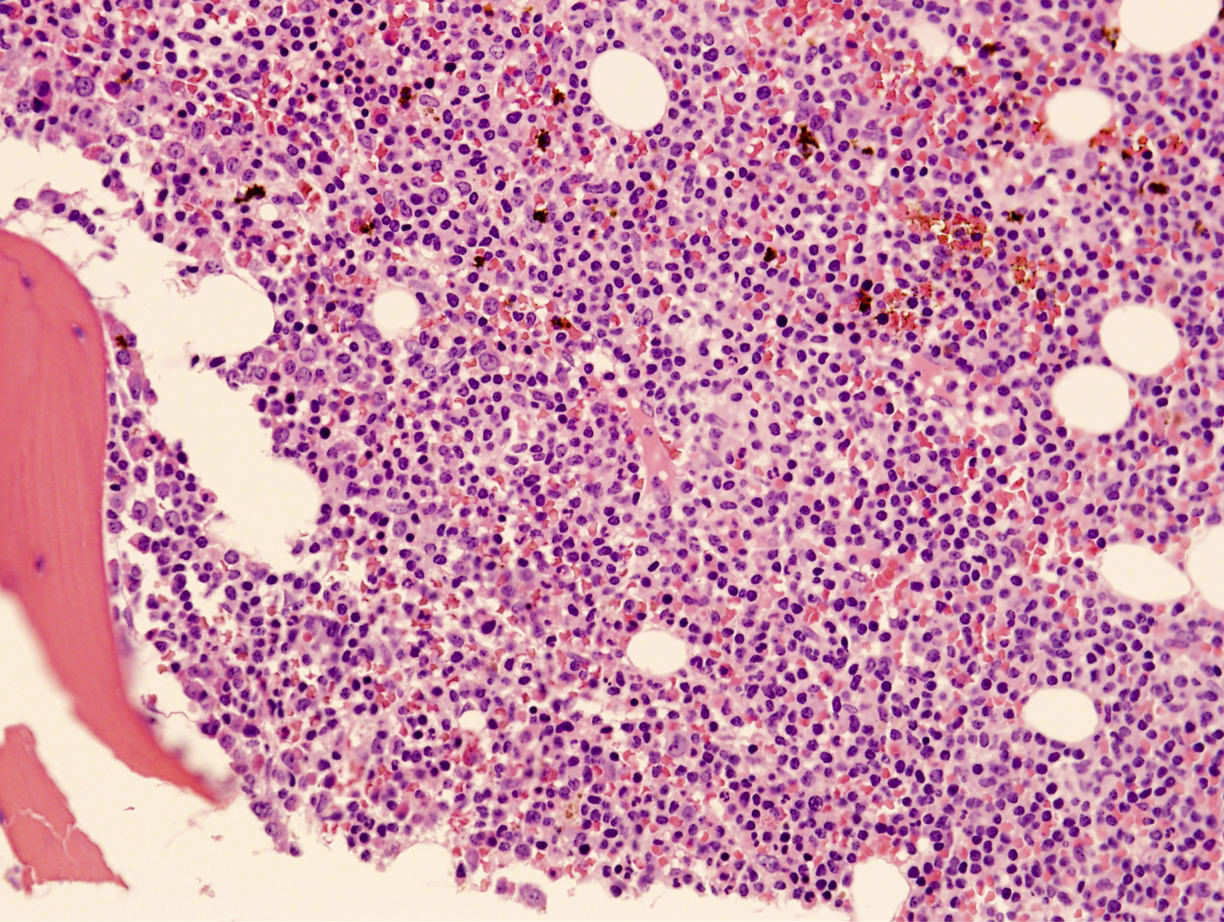
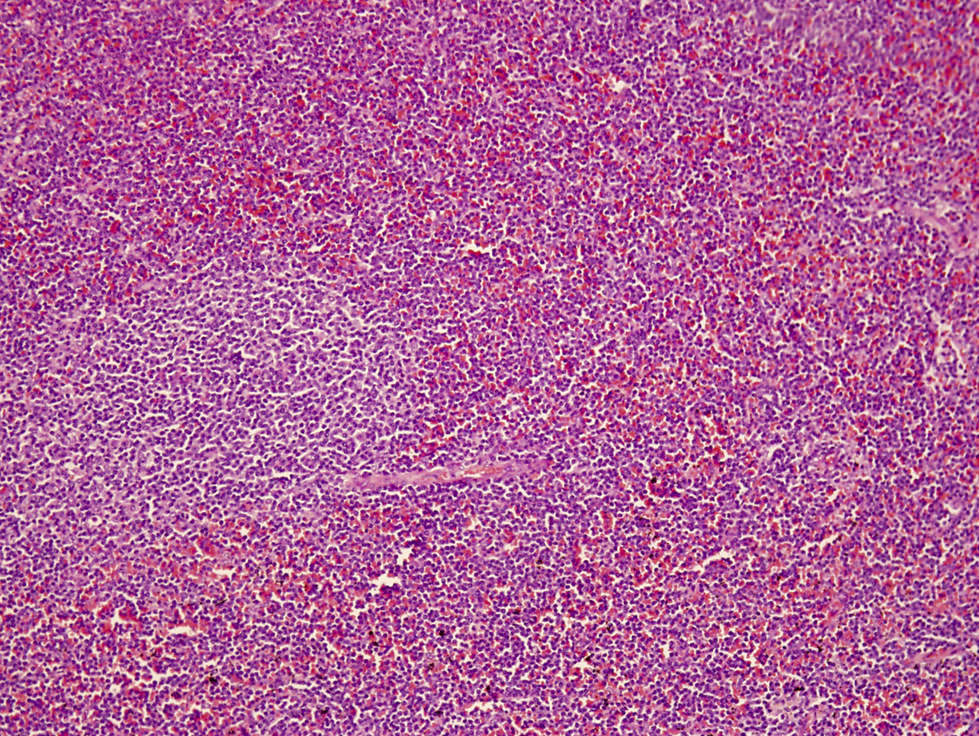
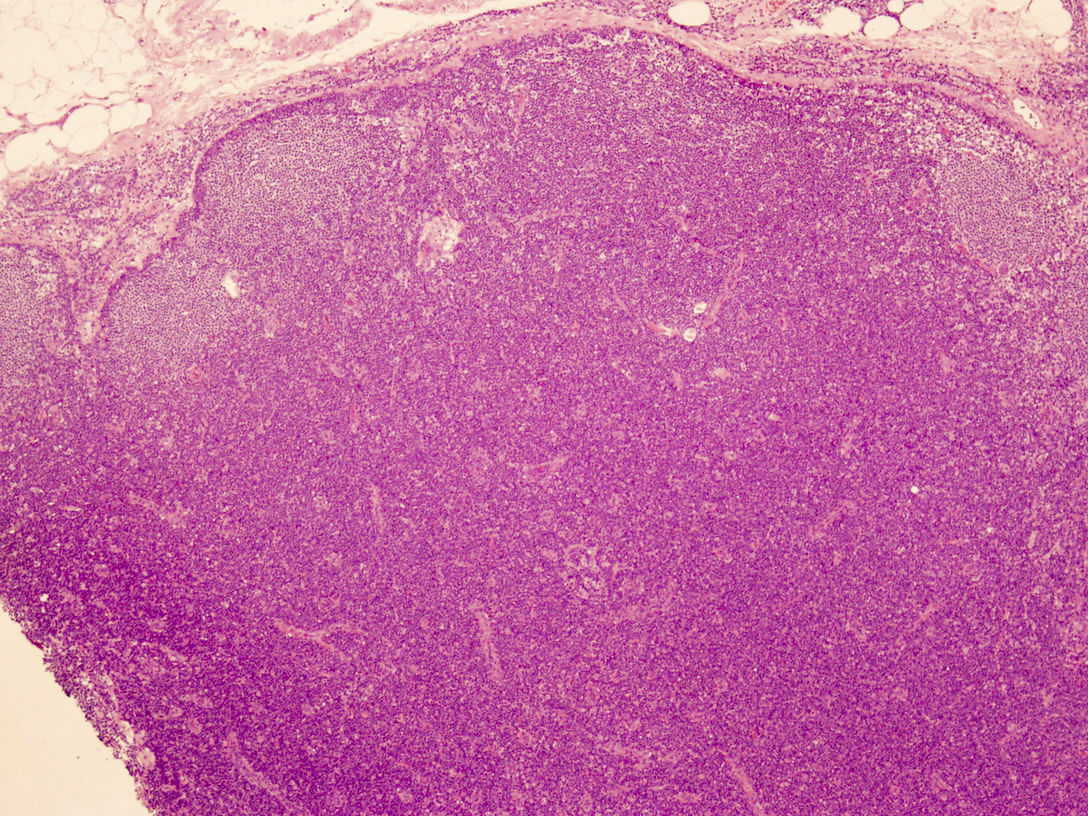
| Hairy cell leukemia
| Hairy cell leukemia
| Hairy cell leukemia
|
| Hairy cell leukemia variant*
| Splenic B cell lymphoma / leukemia with prominent nucleoli (distinct from HCL and includes all previous CD5 negative B PLL)
| Hairy cell leukemia variant*
|
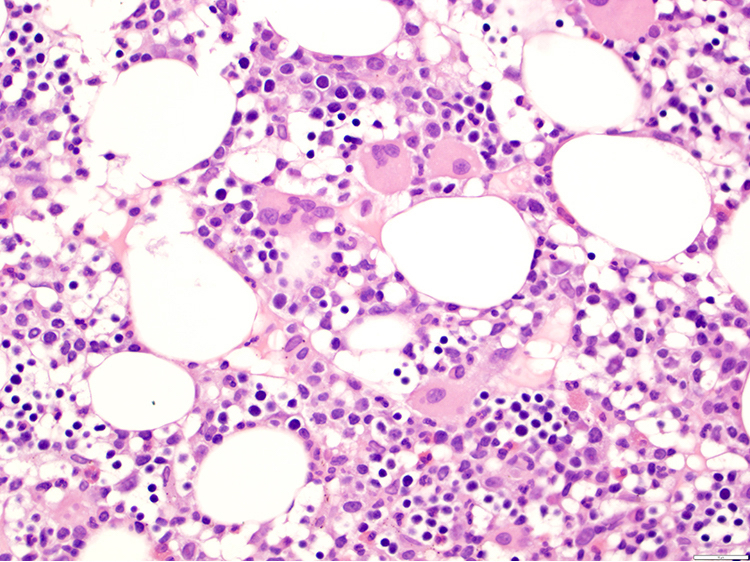
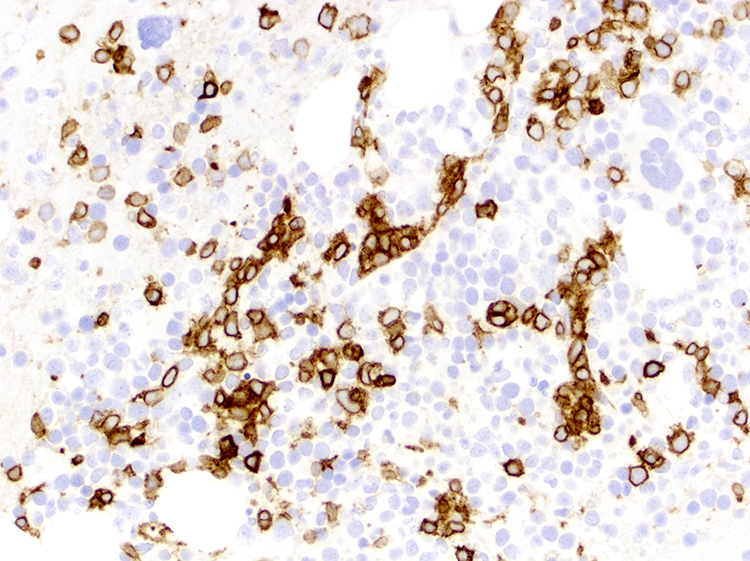
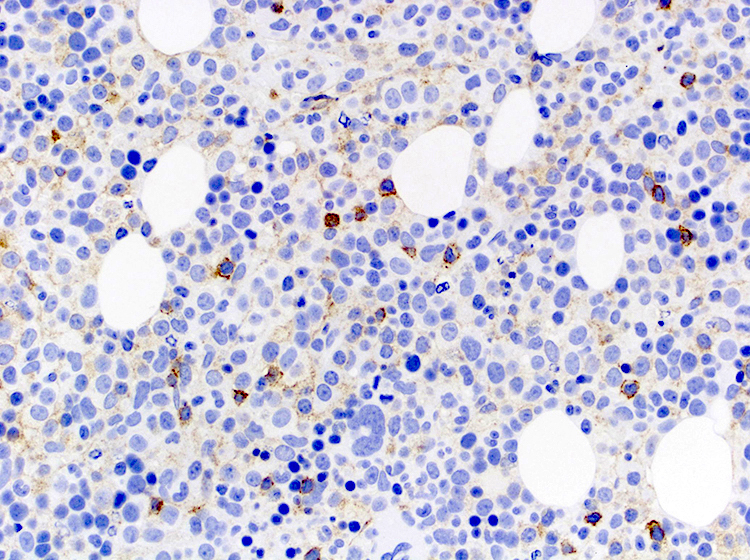
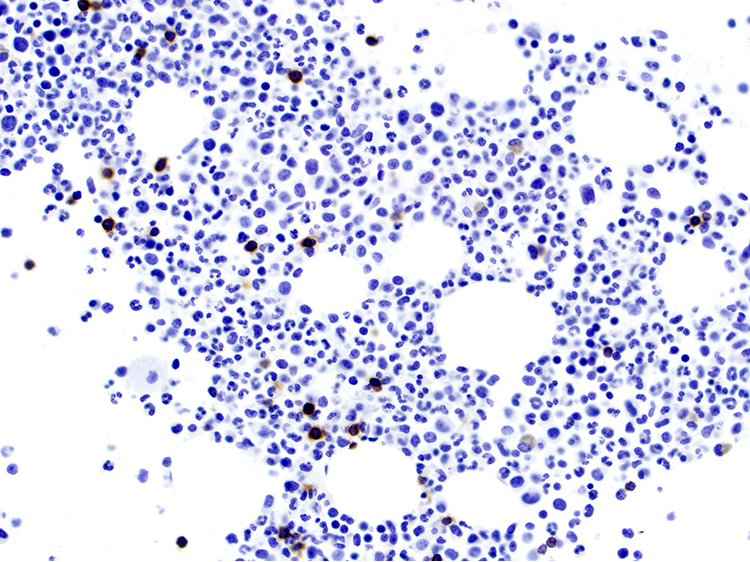
| Splenic diffuse red pulp small B cell lymphoma*
| Splenic diffuse red pulp small B cell lymphoma
| Splenic diffuse red pulp small B cell lymphoma*
|
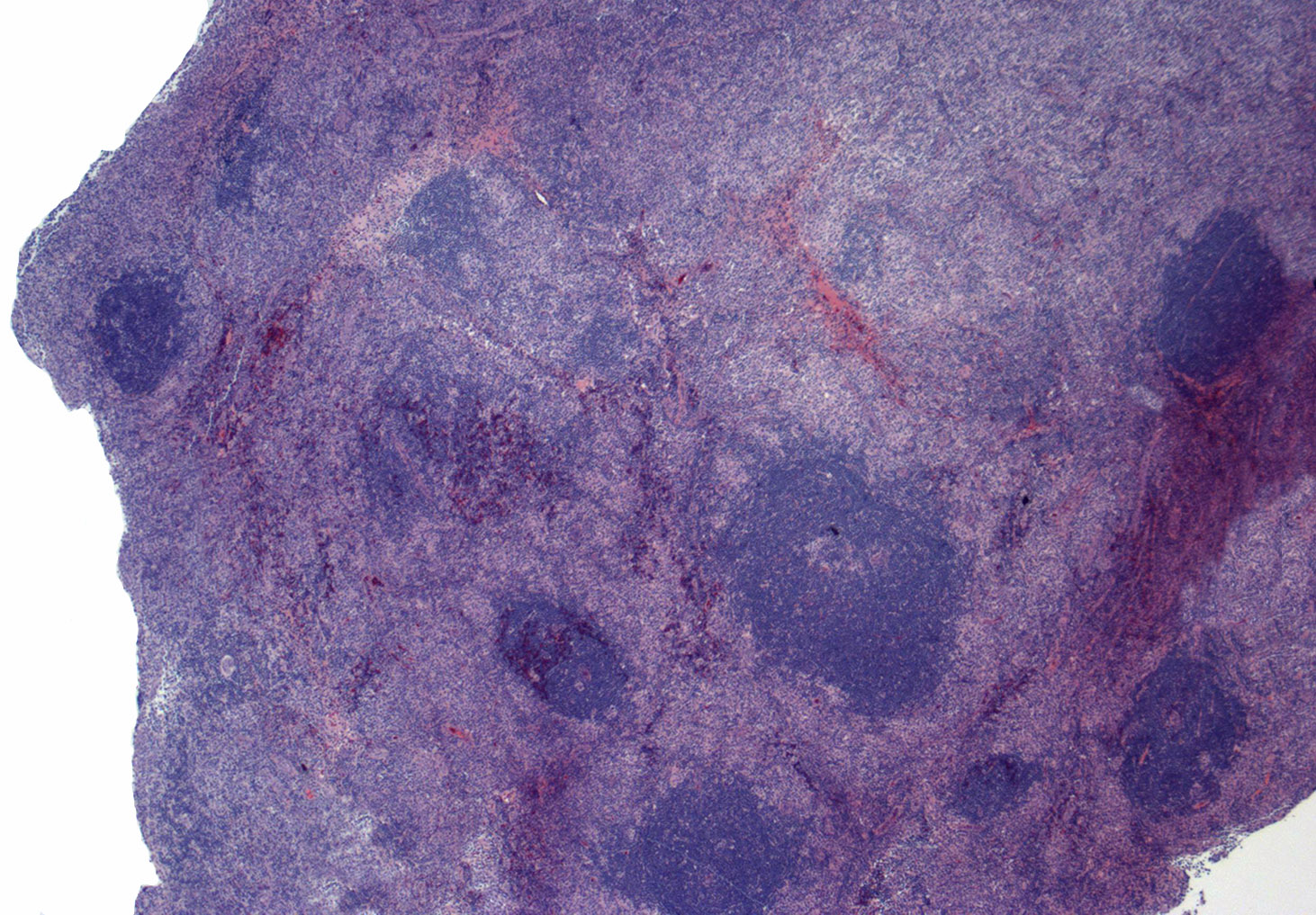
| Follicular lymphoma
|
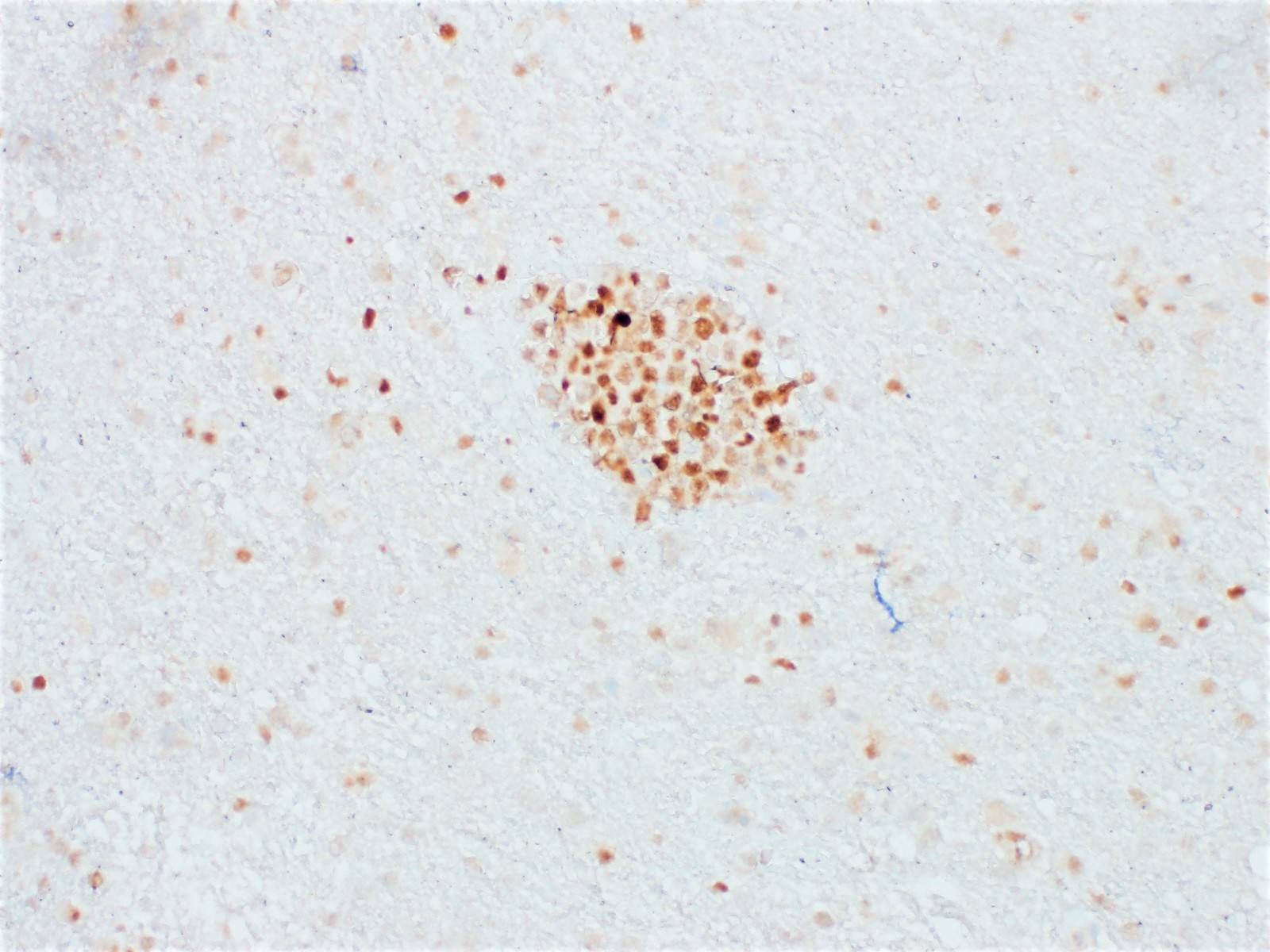
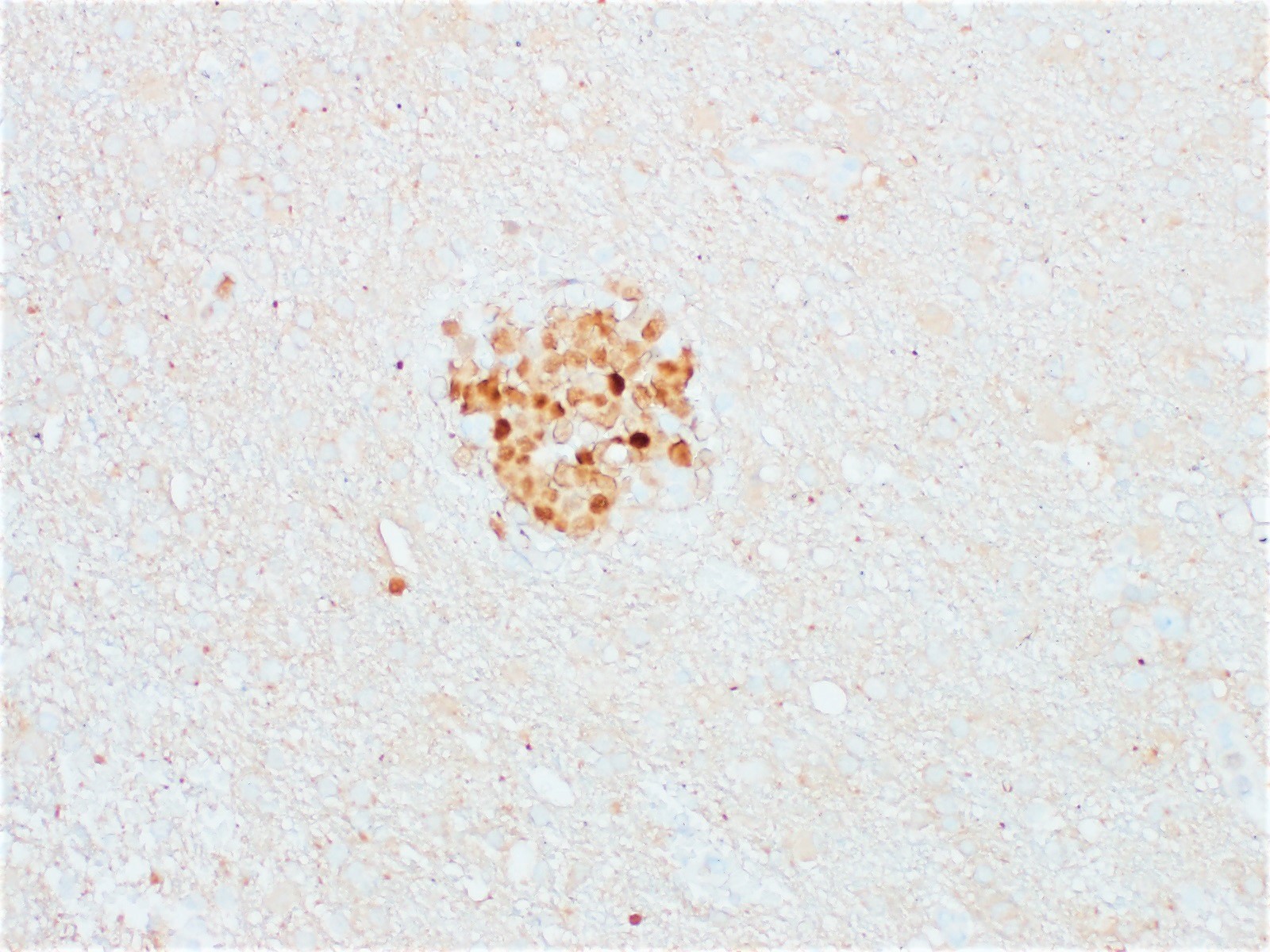
| In situ follicular neoplasia
| In situ follicular B cell neoplasm
| In situ follicular neoplasia
|
| Follicular lymphoma (FL)
| Follicular lymphoma (grading of cFL not mandatory) - Classic follicular lymphoma (cFL)
- Follicular large B cell lymphoma (FLBL)
- Follicular lymphoma with uncommon features (uFL)
| Follicular lymphoma (continue grading FL with emphasis on 3A and 3B) - Follicular lymphoma
- BCL2 rearrangement negative, CD23 positive follicle center lymphoma*
|
| Pediatric type follicular lymphoma
| Pediatric type follicular lymphoma
| Pediatric type follicular lymphoma
|
| Not included
|
| Testicular follicular lymphoma
|
| Duodenal type follicular lymphoma
| Duodenal type follicular lymphoma
| Duodenal type follicular lymphoma
|
| Primary cutaneous follicle center lymphoma
| Primary cutaneous follicle center lymphoma
| Primary cutaneous follicle center lymphoma
|
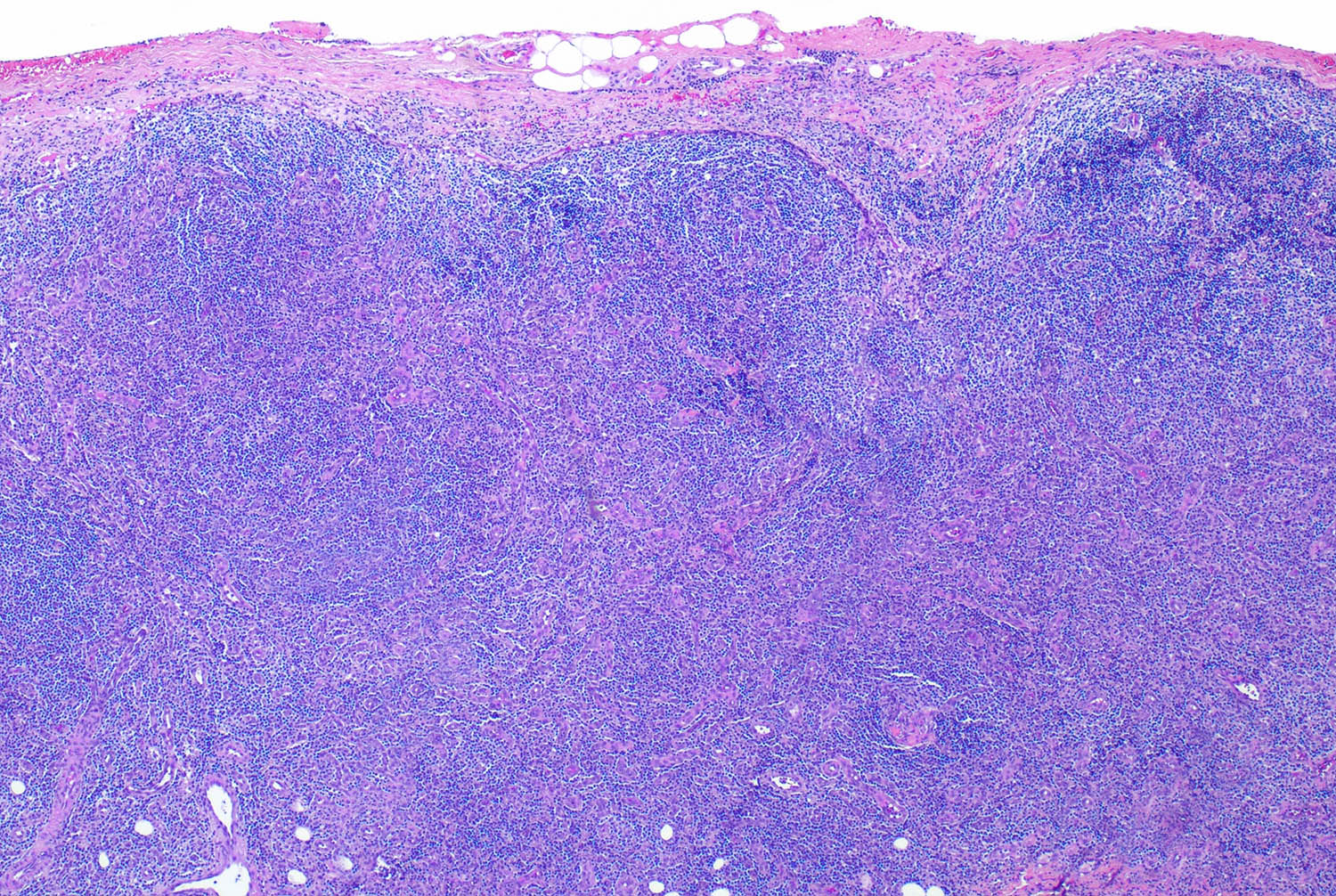
| Mantle cell lymphoma
|
| In situ mantle cell neoplasia
| In situ mantle cell neoplasia
| In situ mantle cell neoplasia
|
| Mantle cell lymphoma
| Mantle cell lymphoma
| Mantle cell lymphoma
|
| Leukemic nonnodal mantle cell lymphoma
| Leukemic nonnodal mantle cell lymphoma
| Leukemic nonnodal mantle cell lymphoma
|
| Aggressive lymphomas transformed from low grade B cell lymphomas
|
| Not included
| Transformations of indolent B cell lymphomas
|
|
| Large B cell lymphoma
|
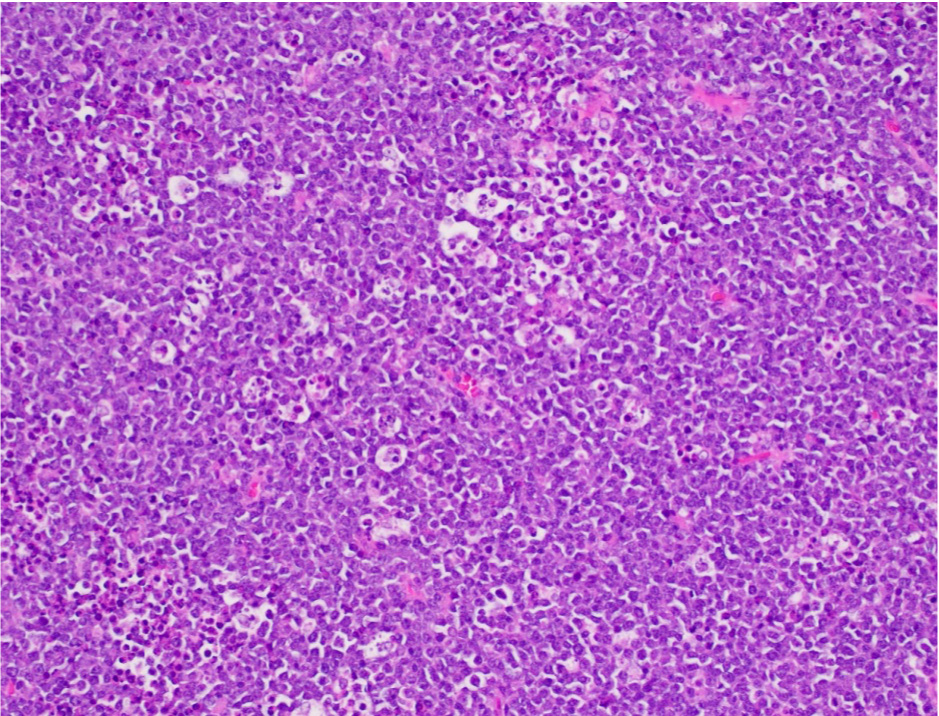
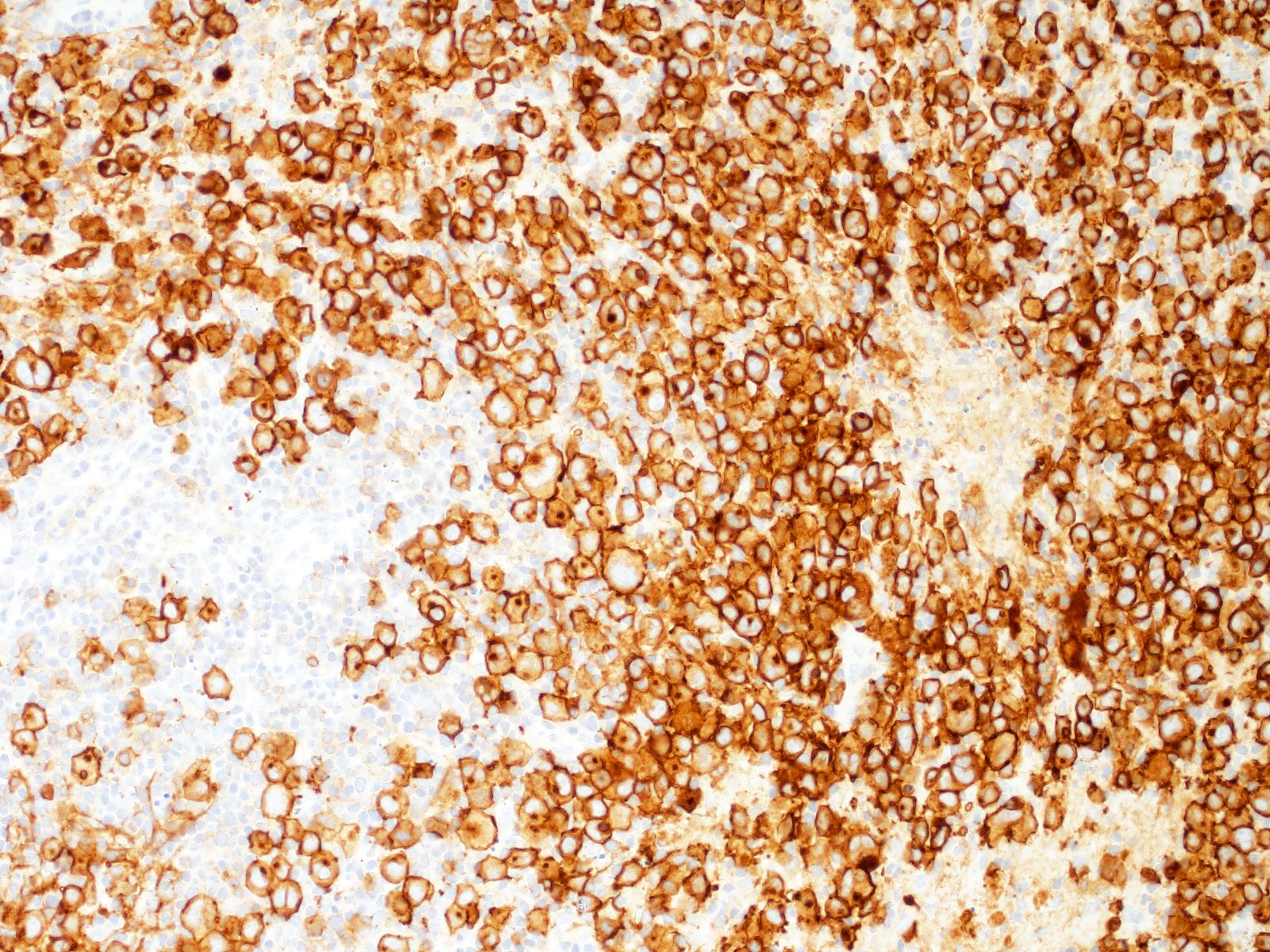
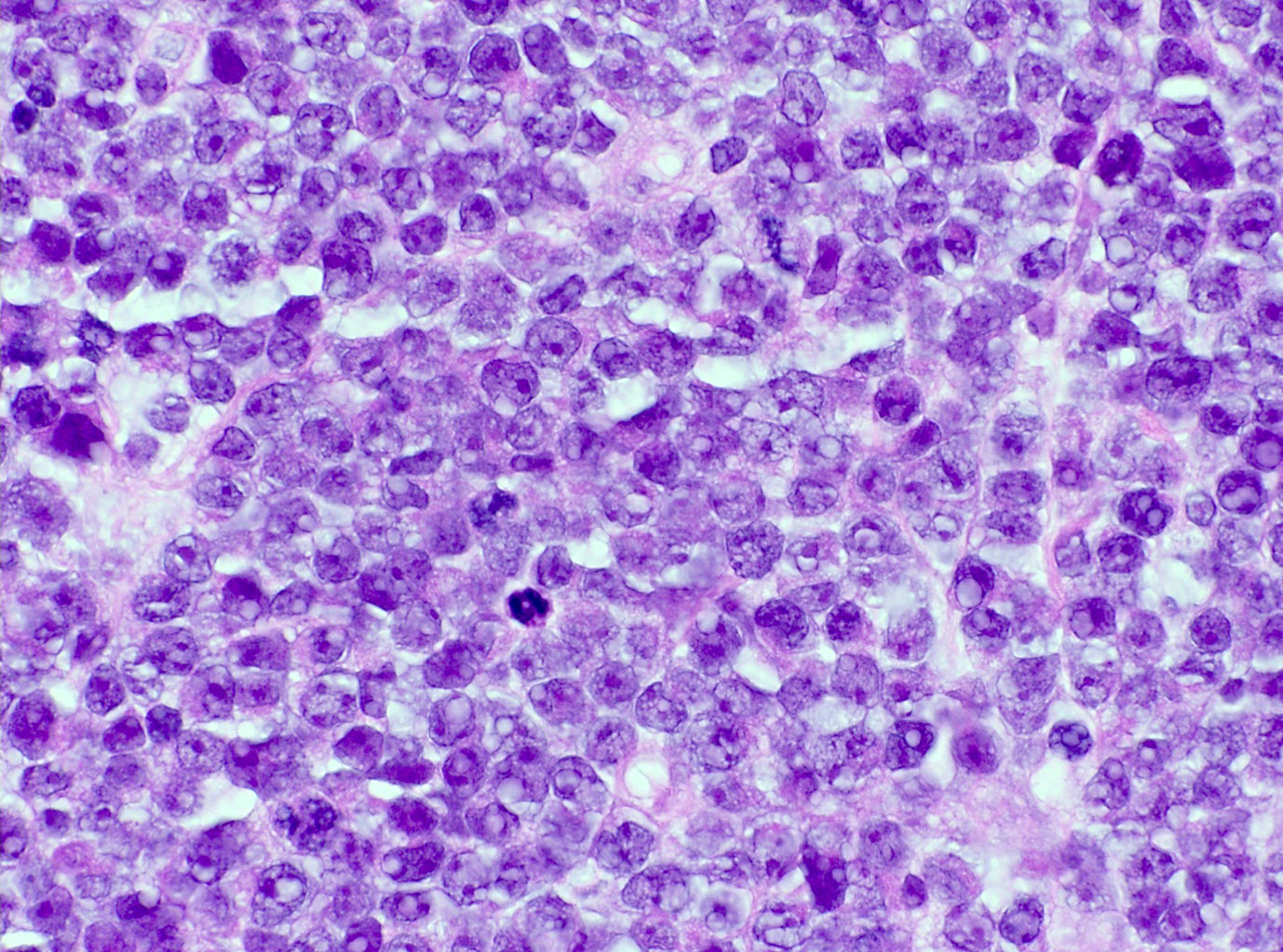
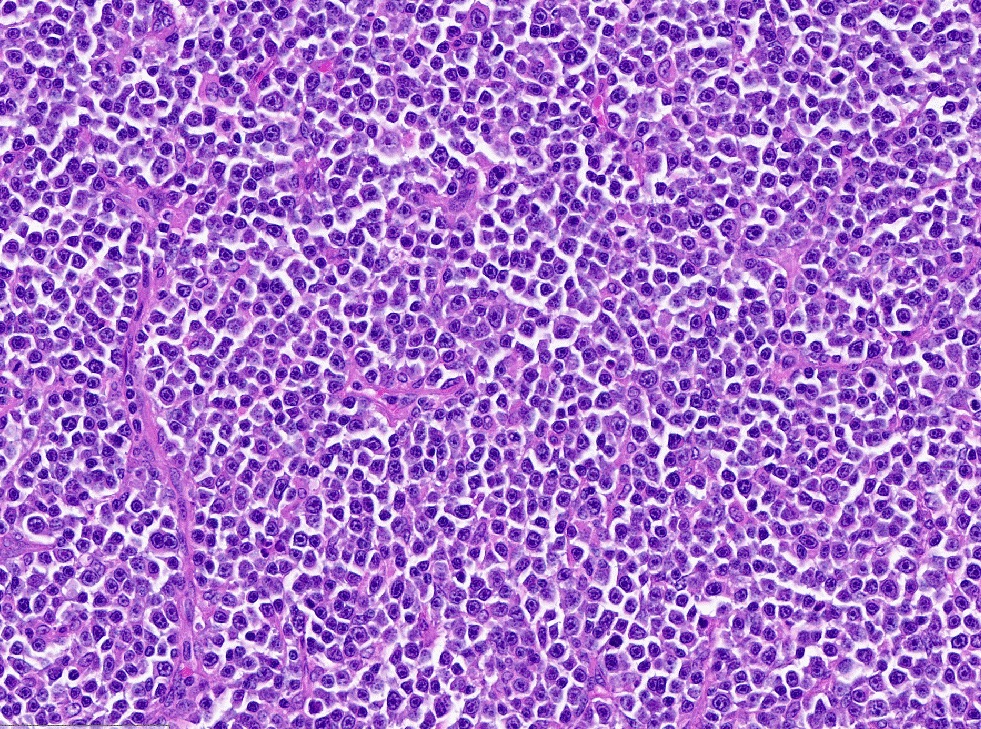
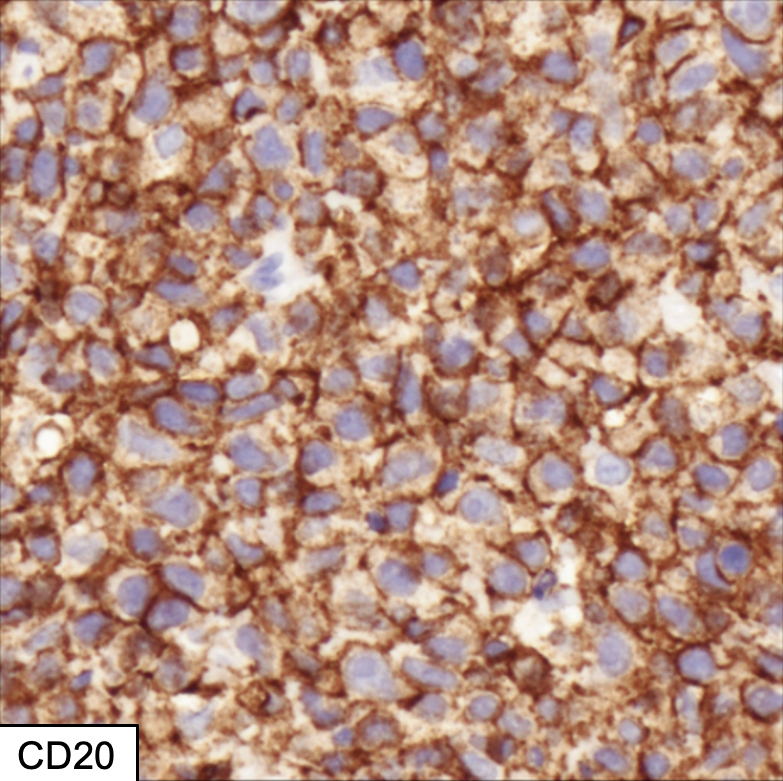
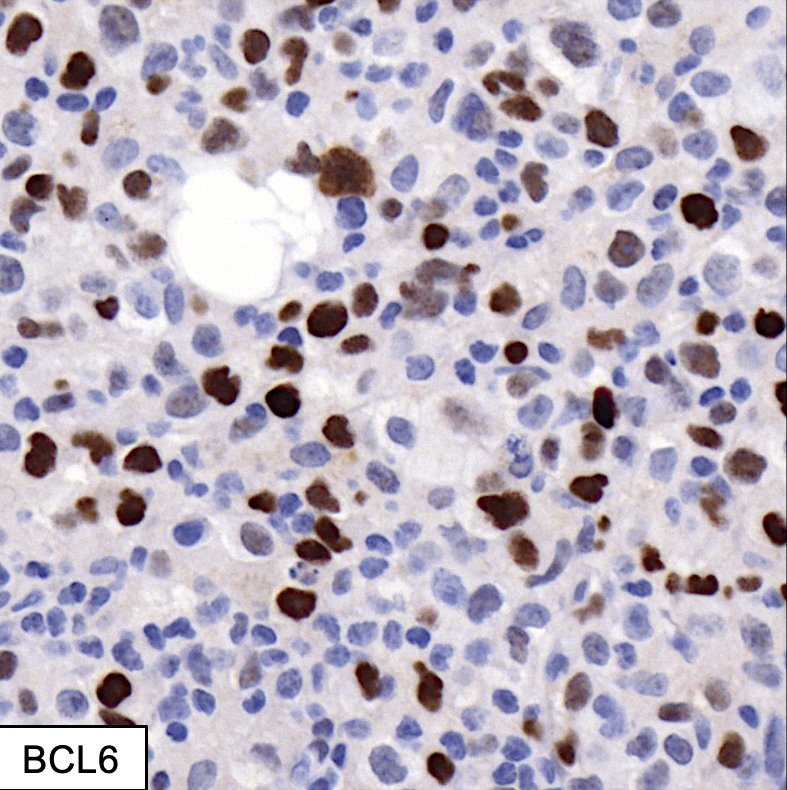
| Diffuse large B cell lymphoma (DLBCL), NOS
| Diffuse large B cell lymphoma, NOS
| Diffuse large B cell lymphoma, NOS
|
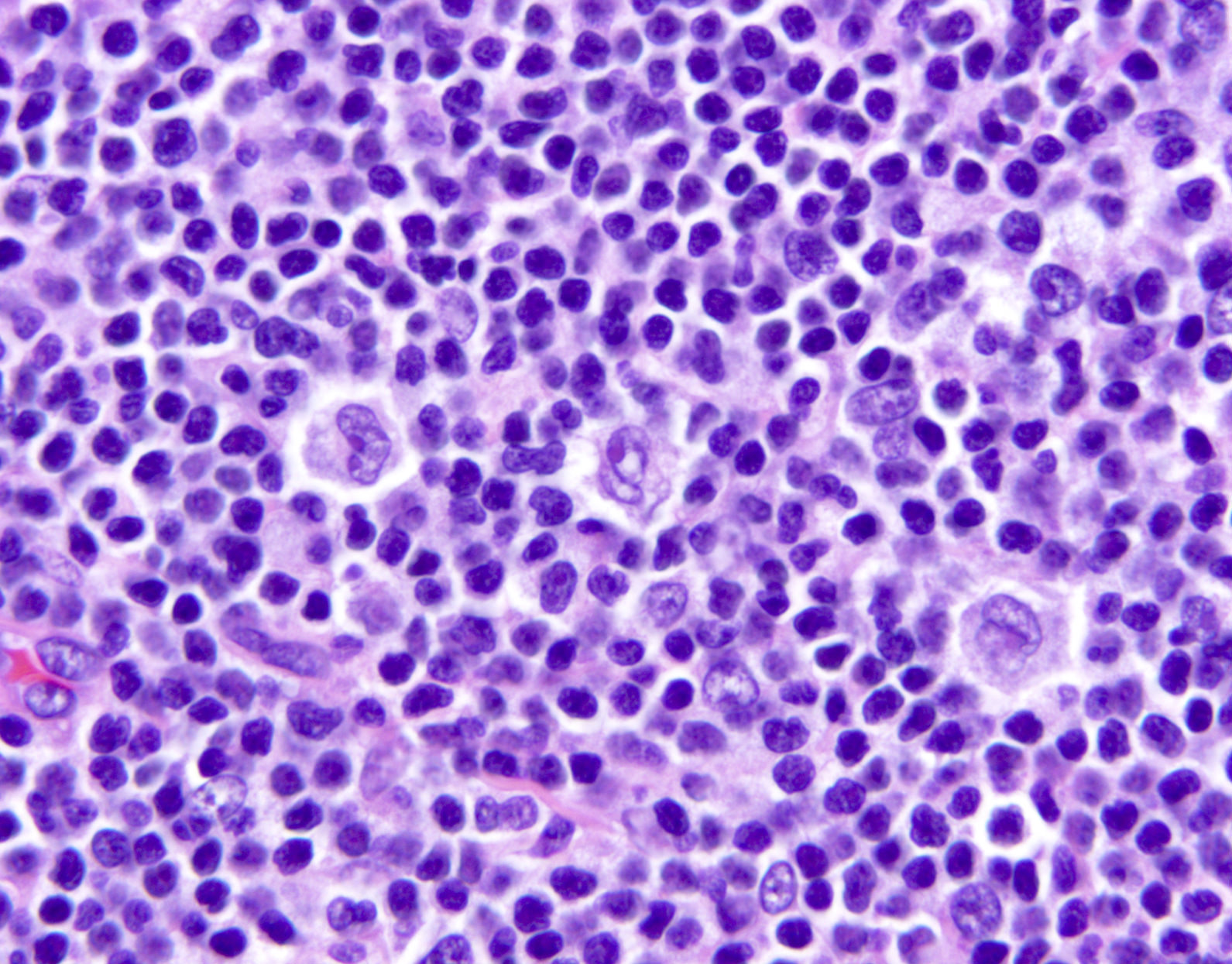
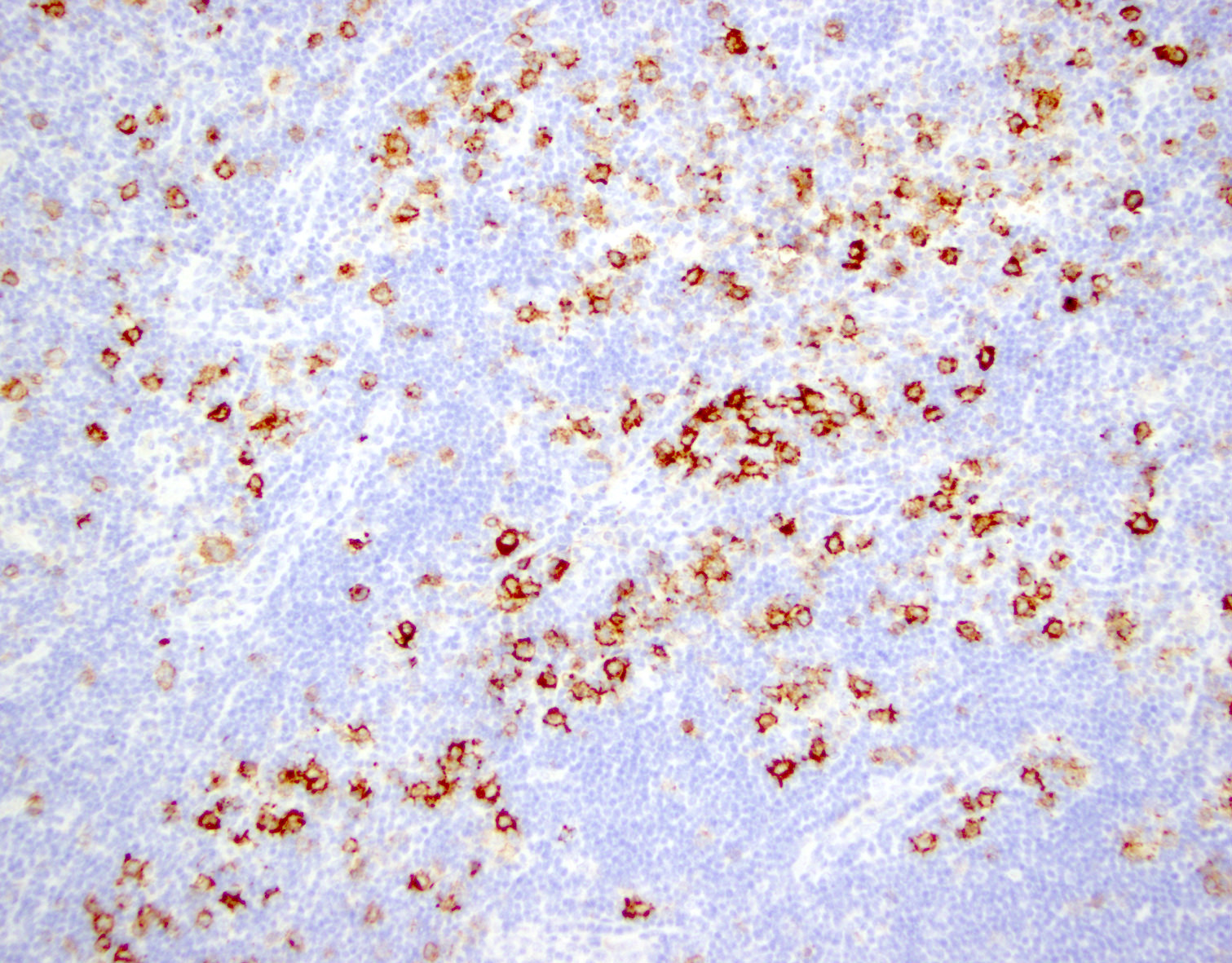
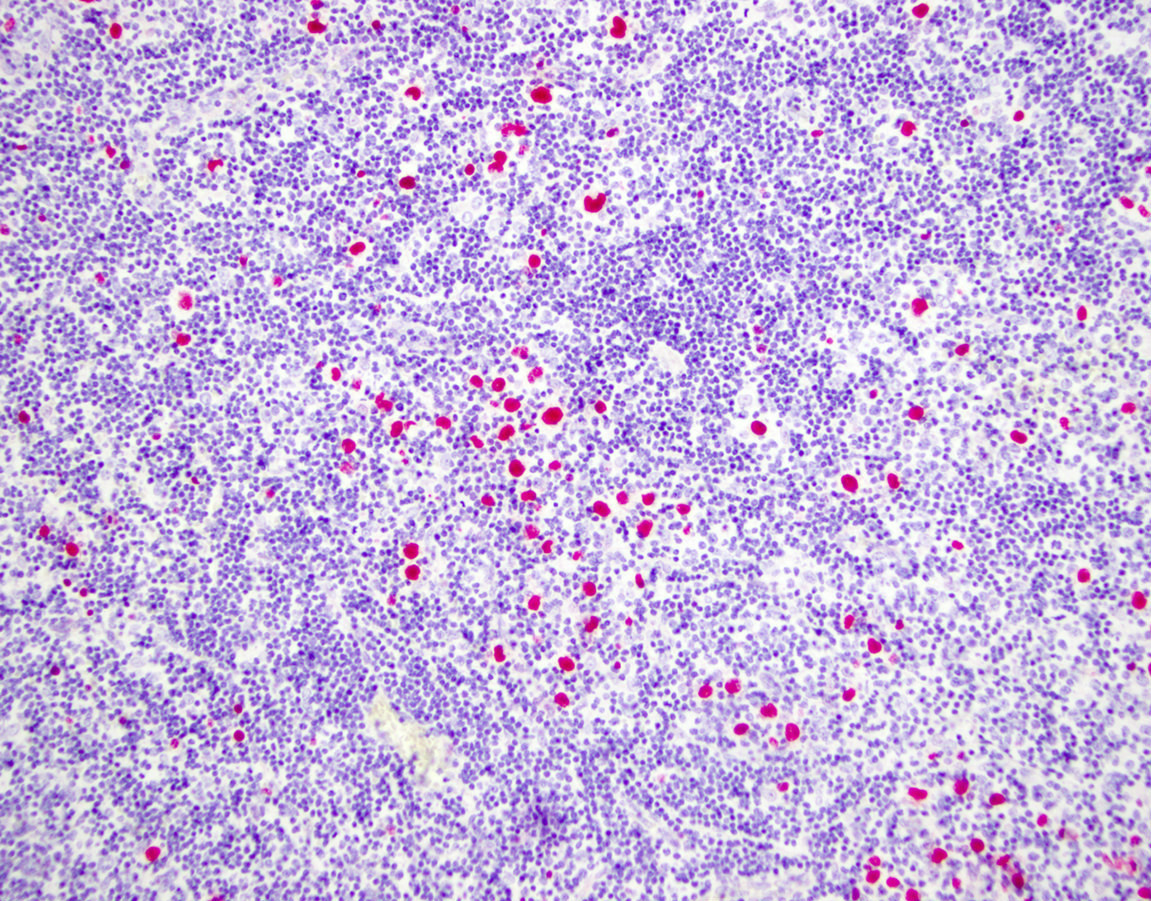
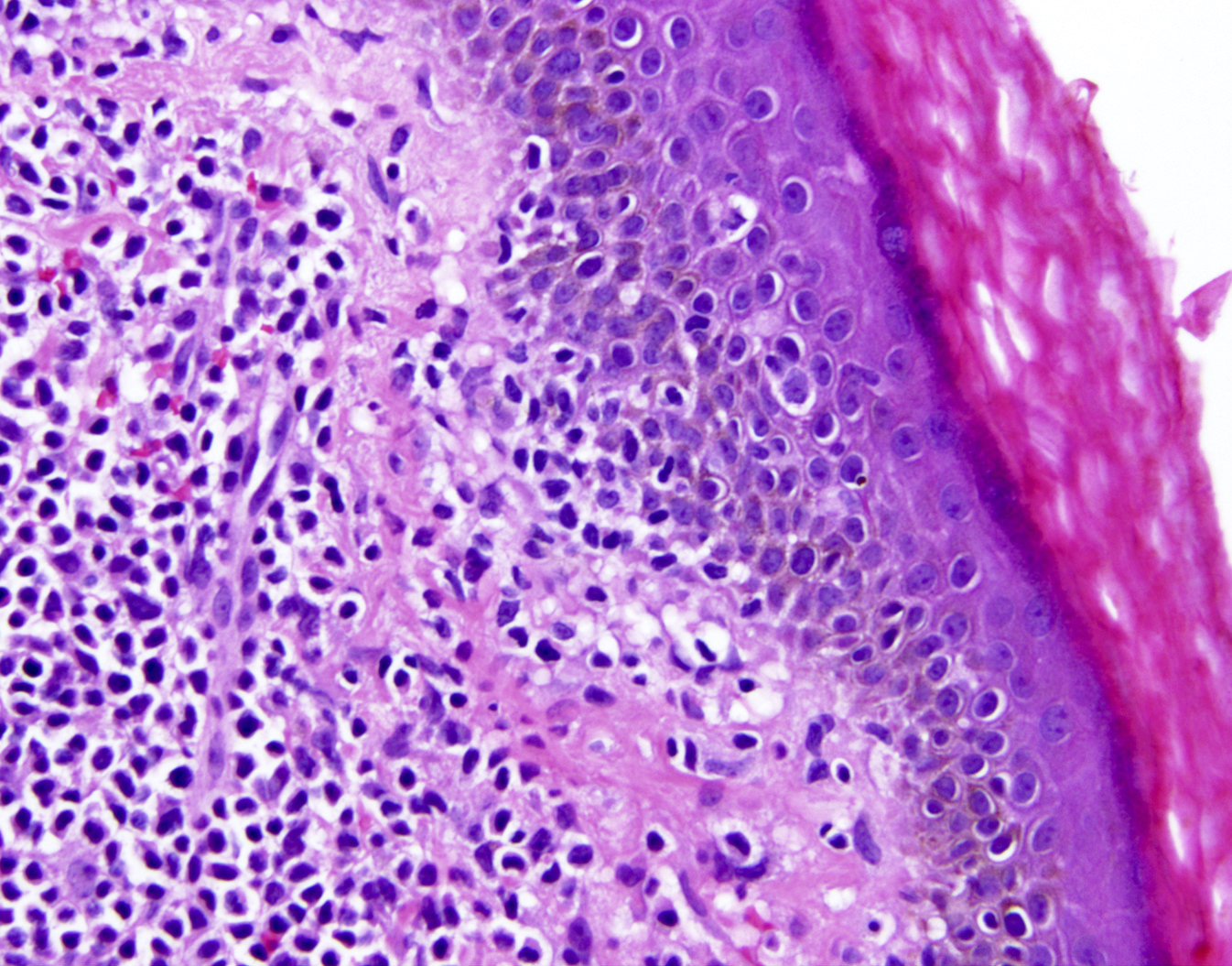
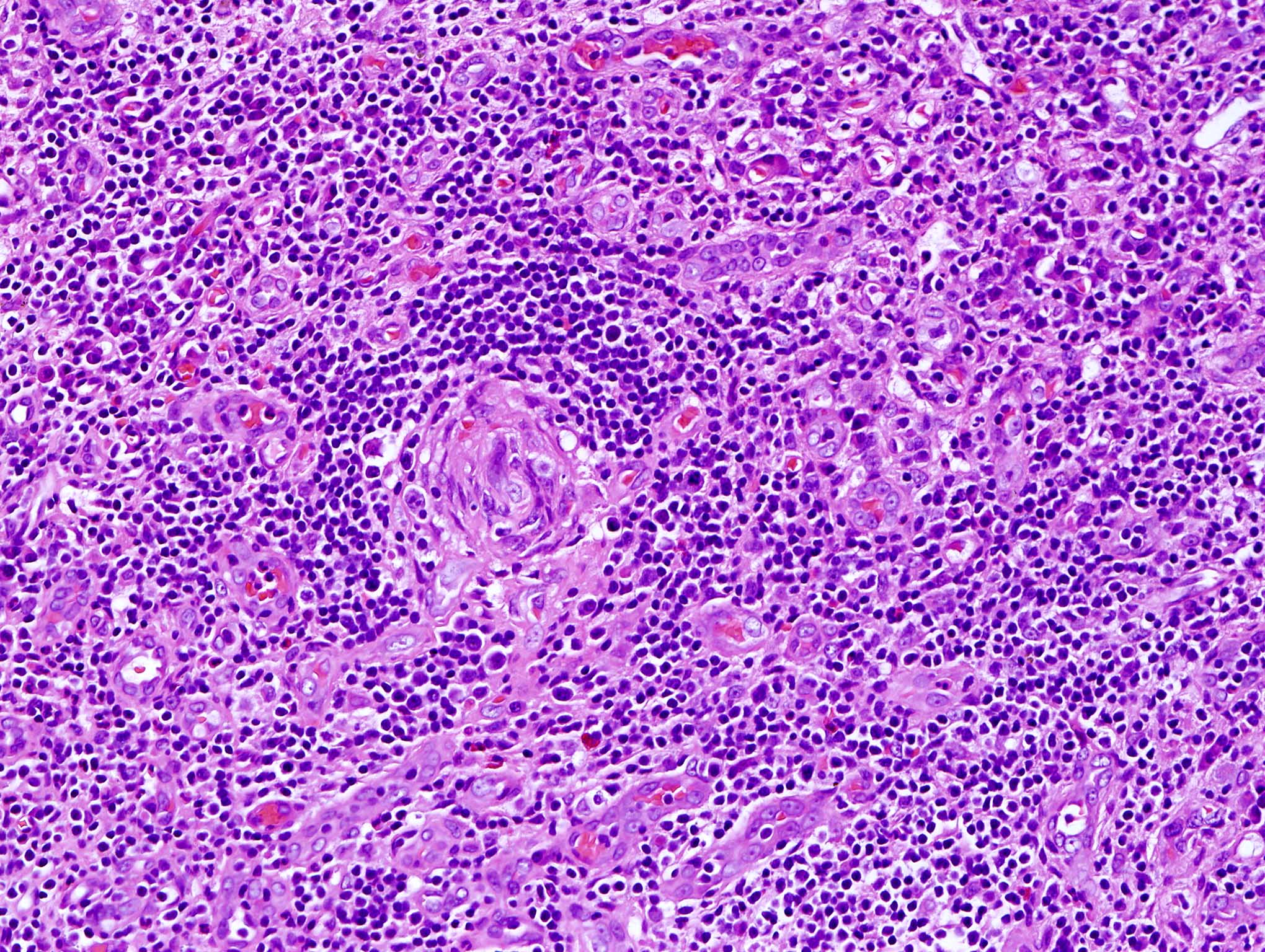
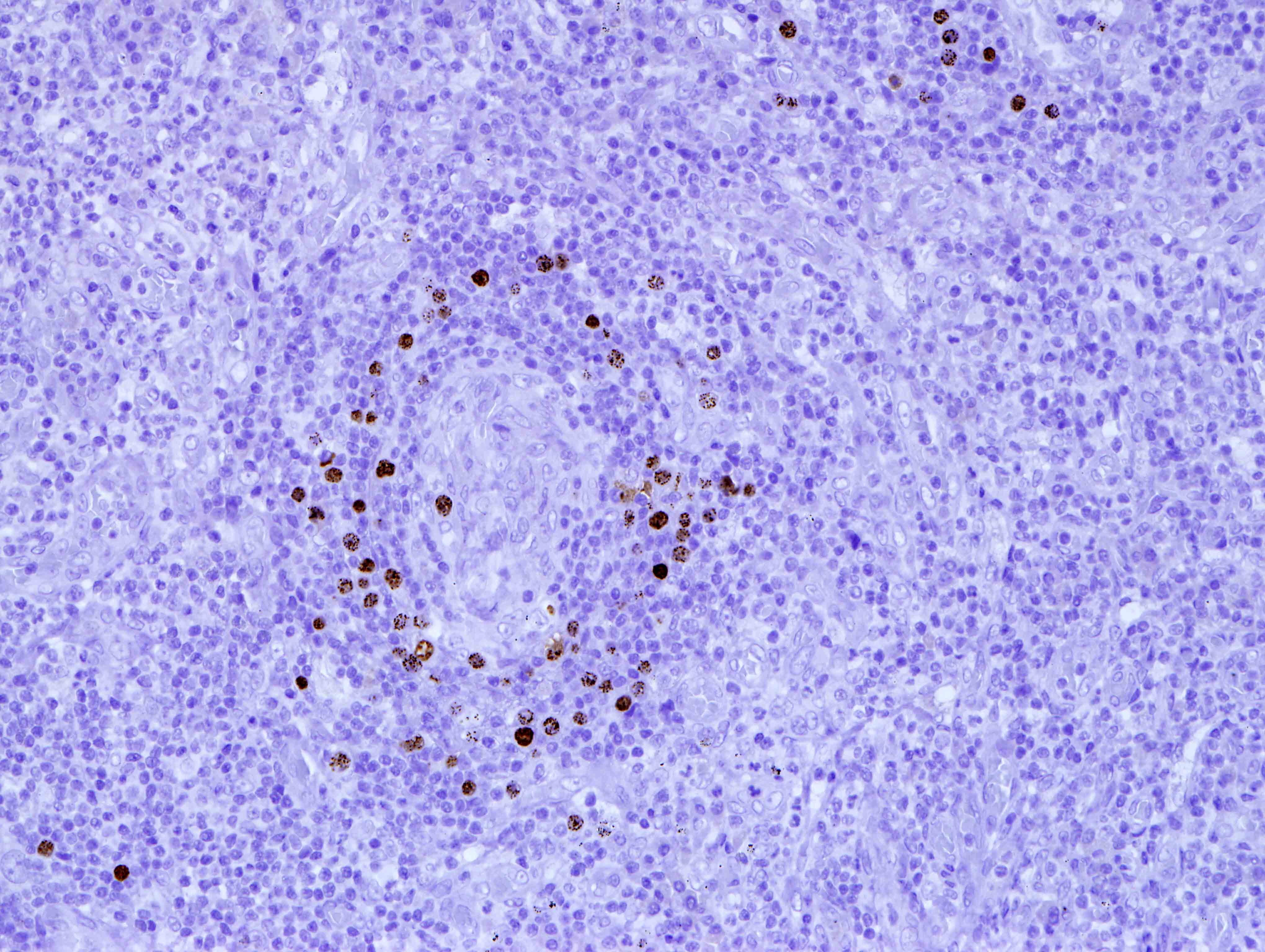
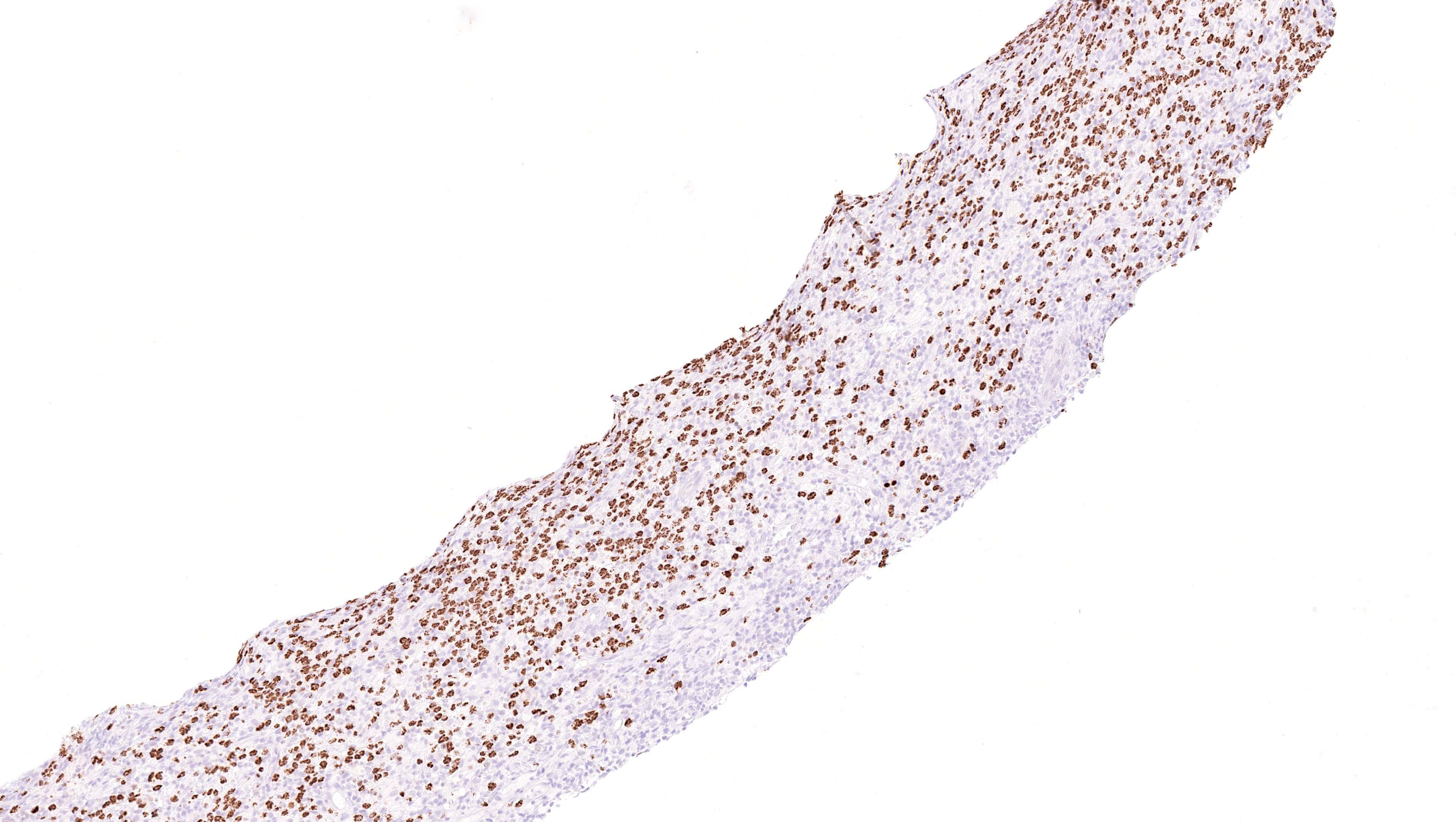
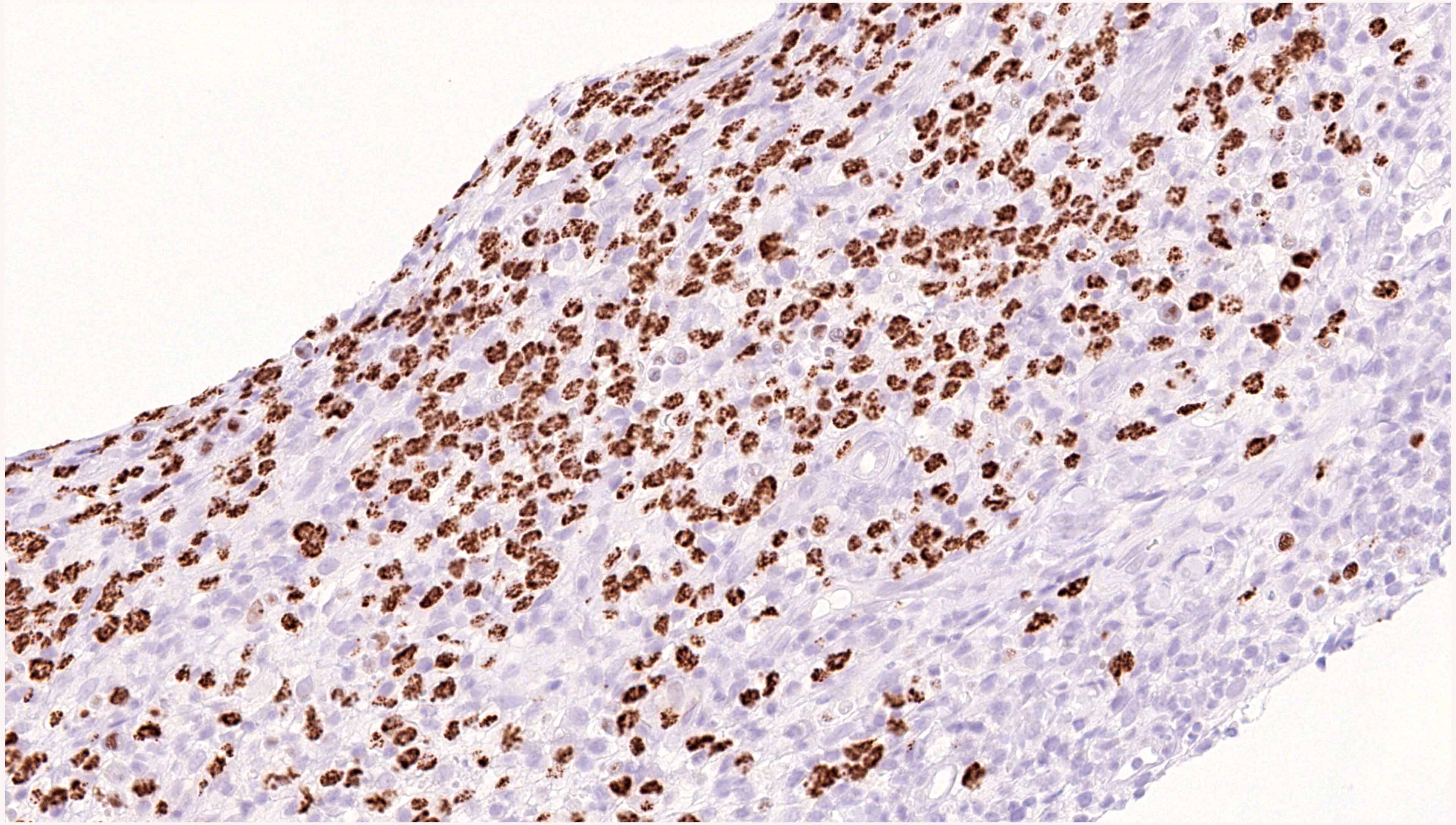
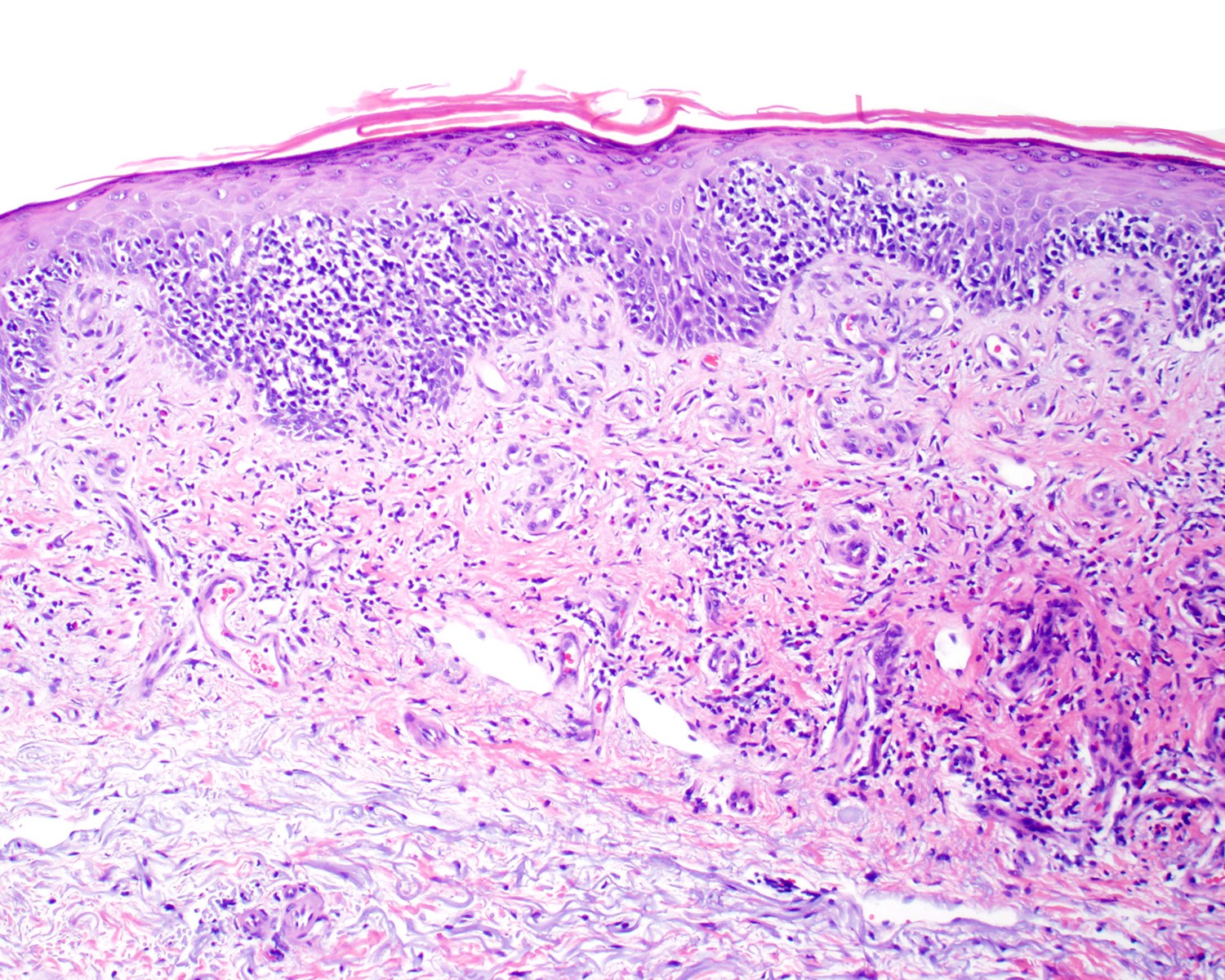
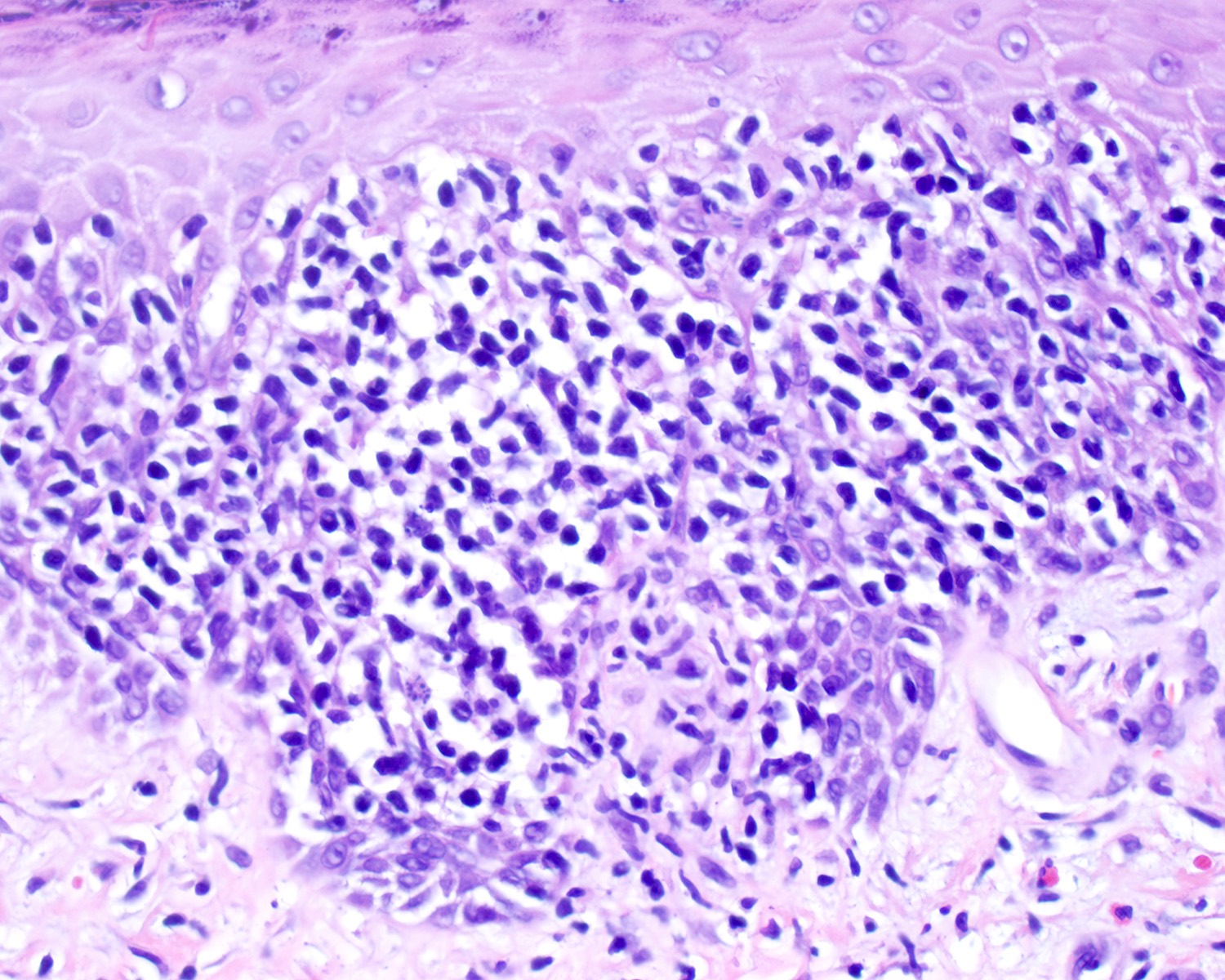
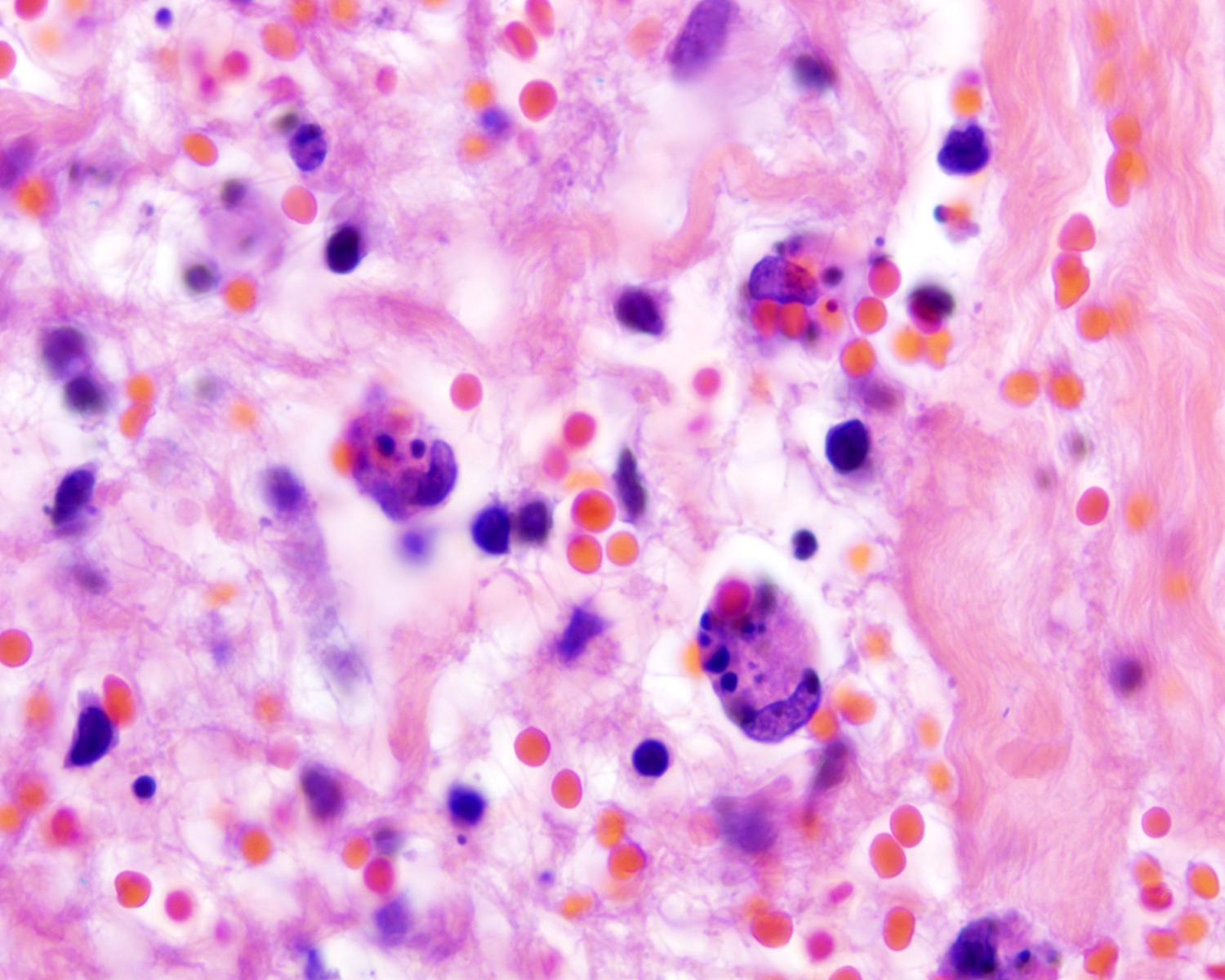
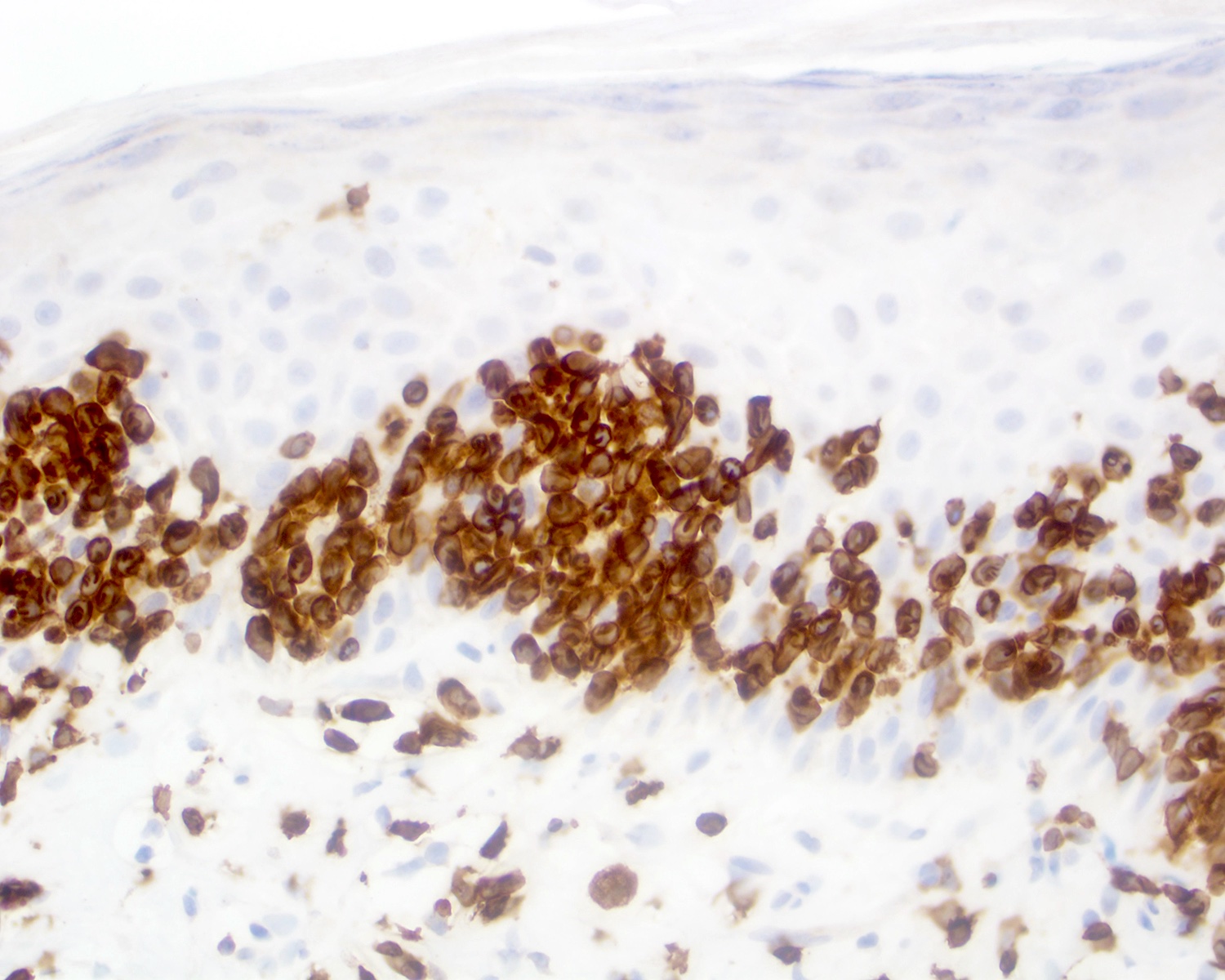
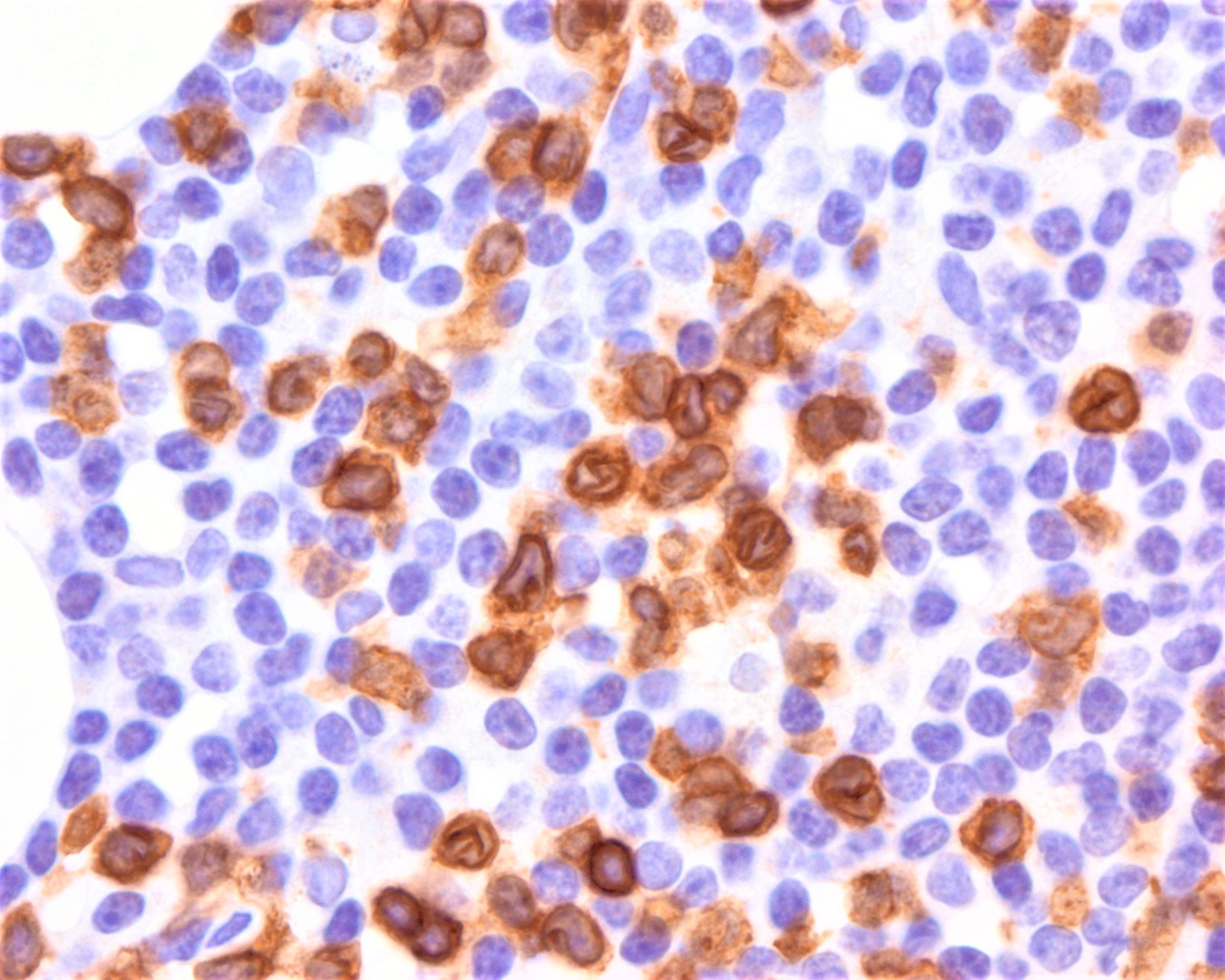
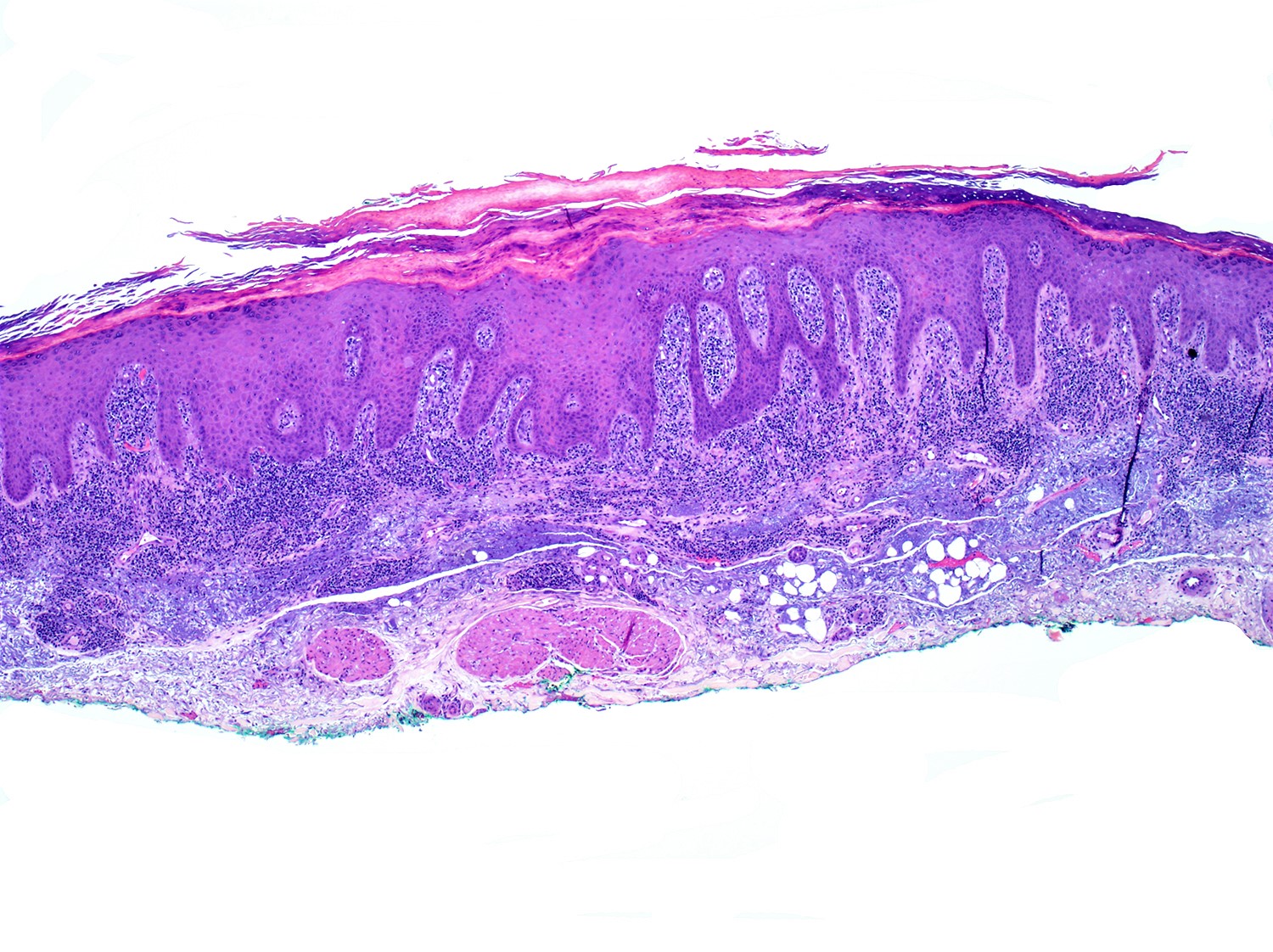
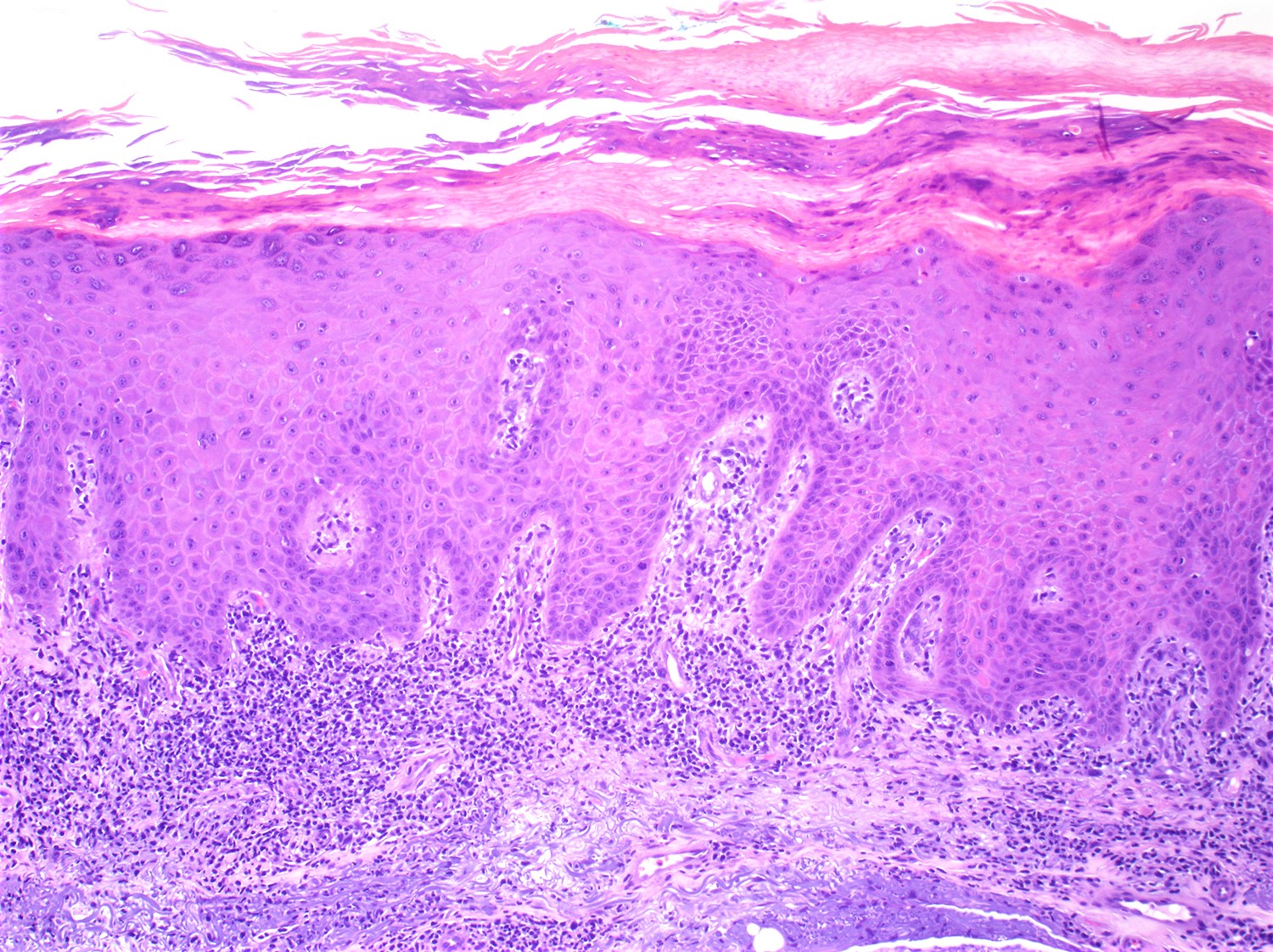
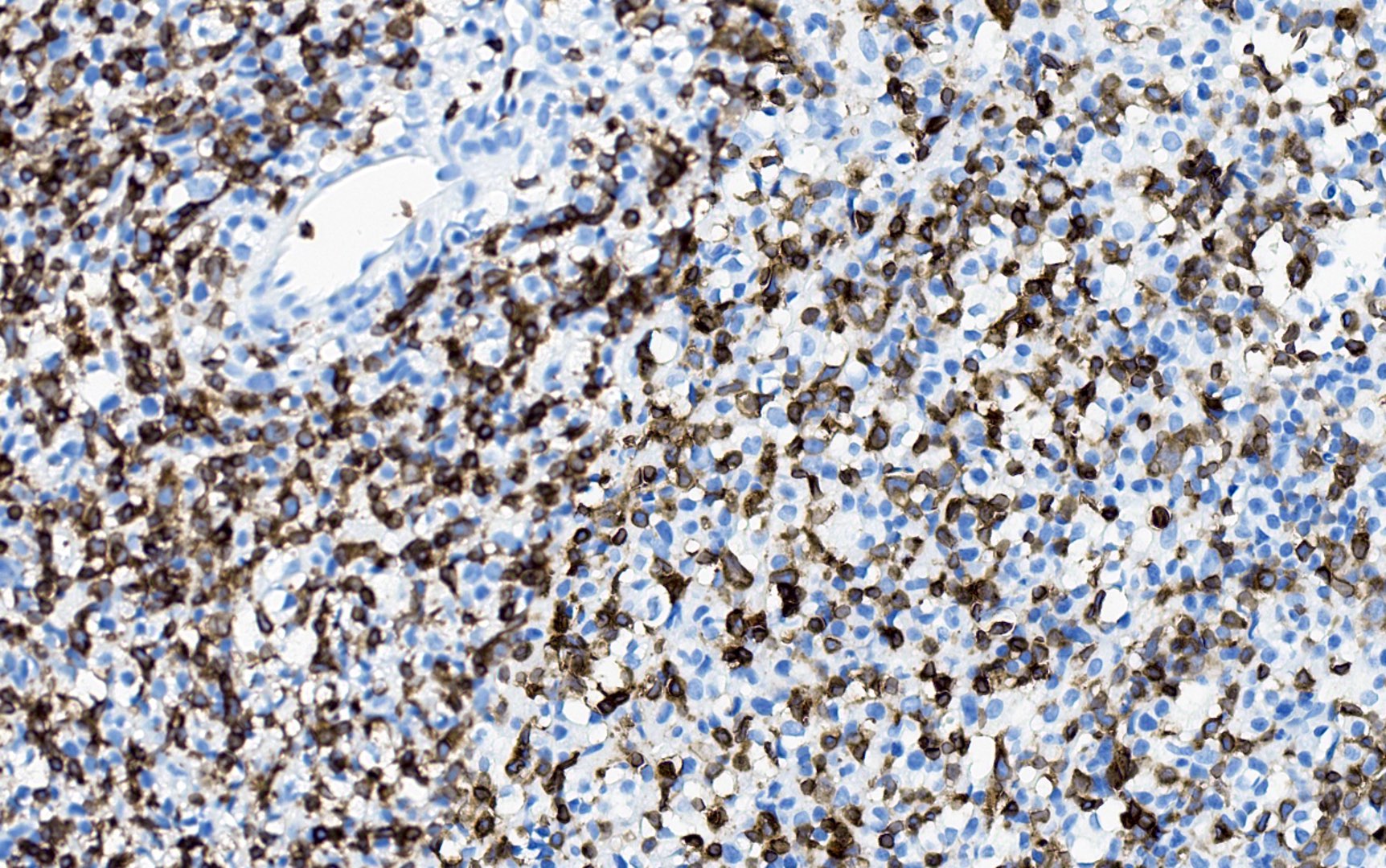
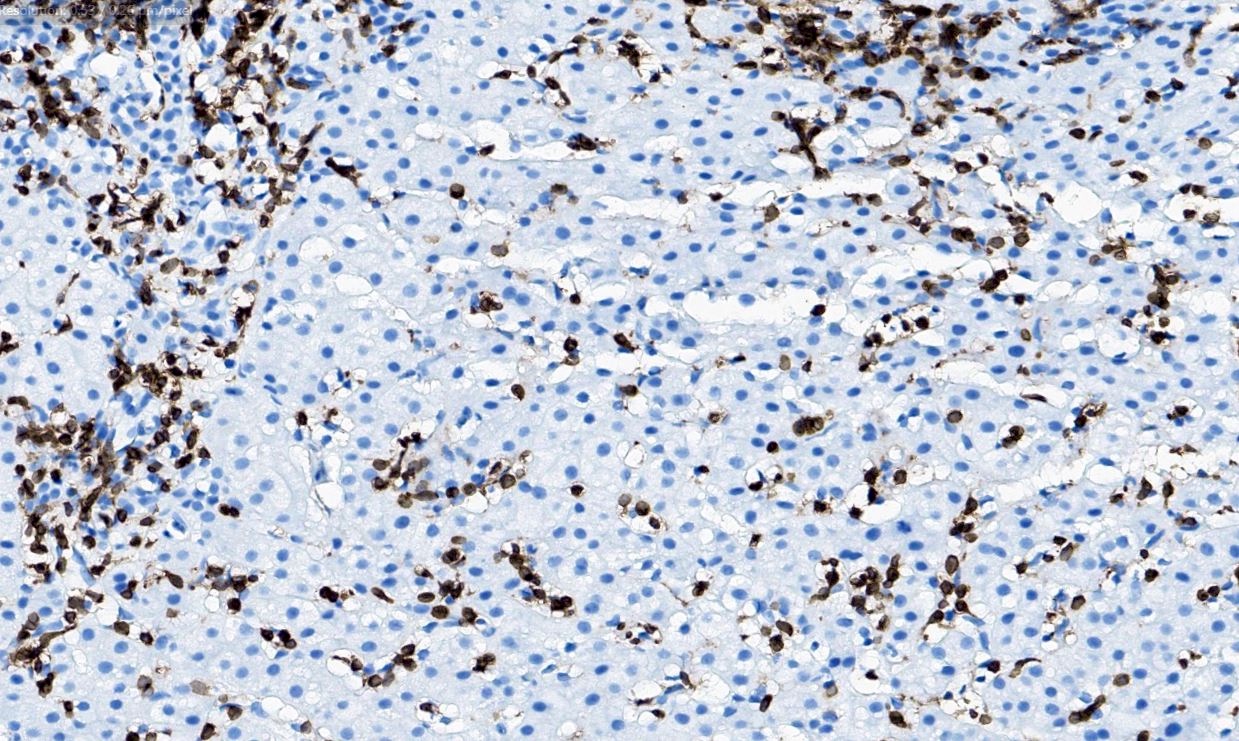
| EBV positive mucocutaneous ulcer*
| EBV positive mucocutaneous ulcer
| EBV positive mucocutaneous ulcer
|
| EBV positive diffuse large B cell lymphoma, NOS
| EBV positive diffuse large B cell lymphoma
| EBV positive diffuse large B cell lymphoma, NOS
|
| Diffuse large B cell lymphoma associated with chronic inflammation
| Diffuse large B cell lymphoma associated with chronic inflammation
| Diffuse large B cell lymphoma associated with chronic inflammation
|
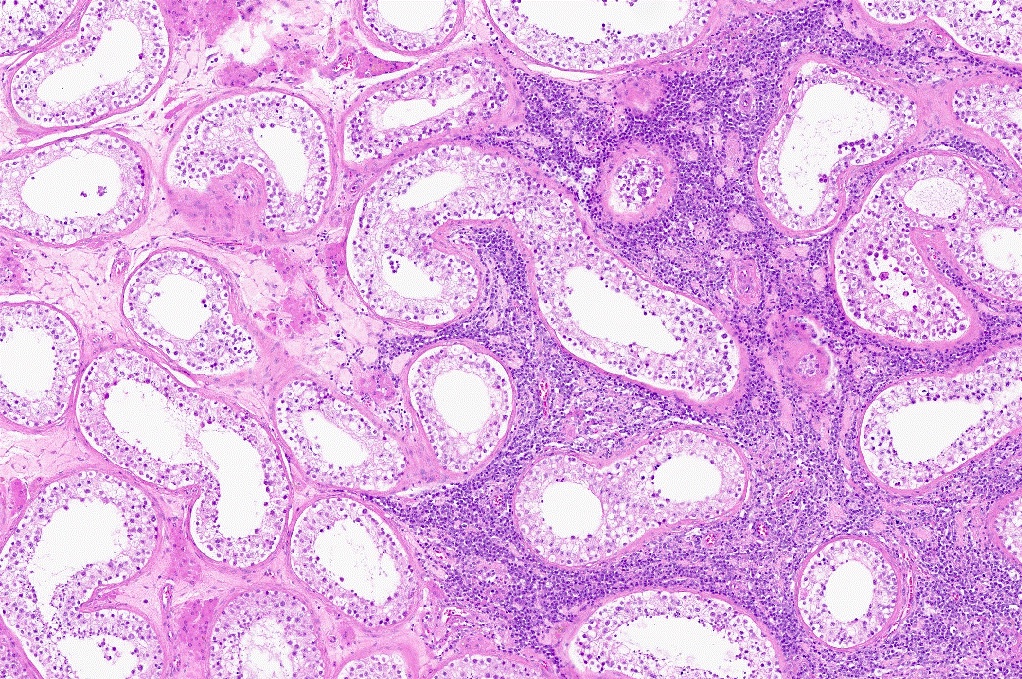
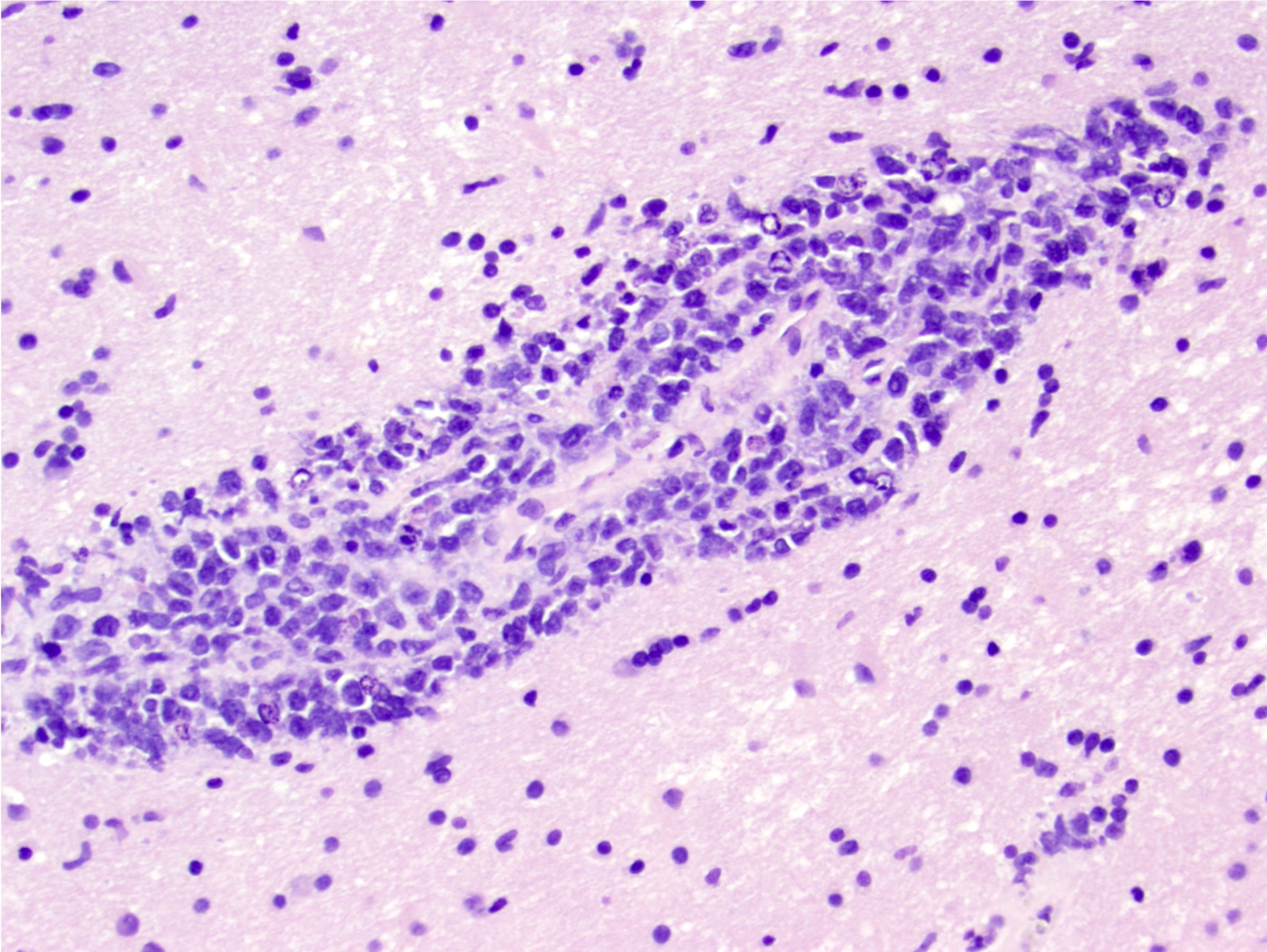
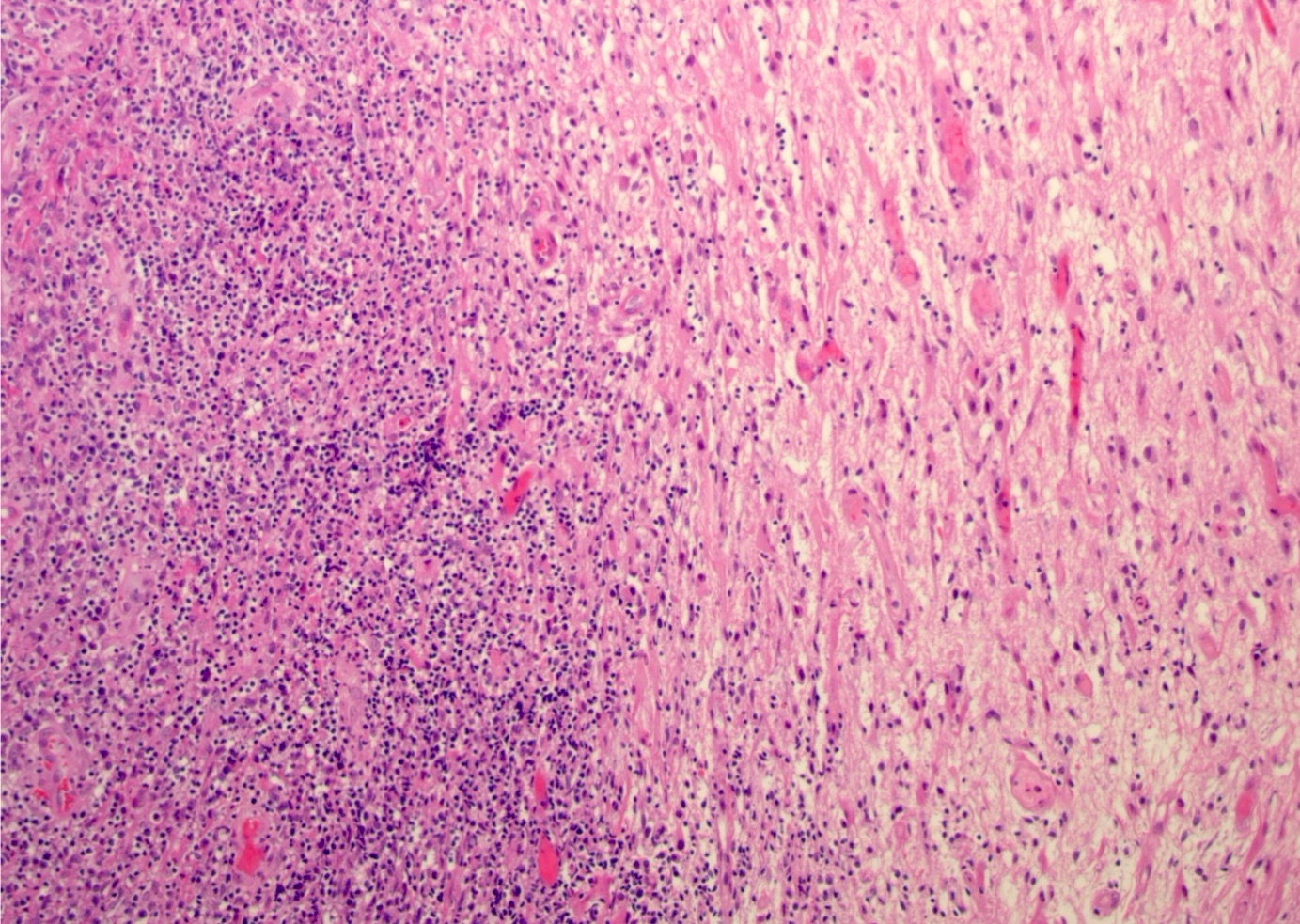
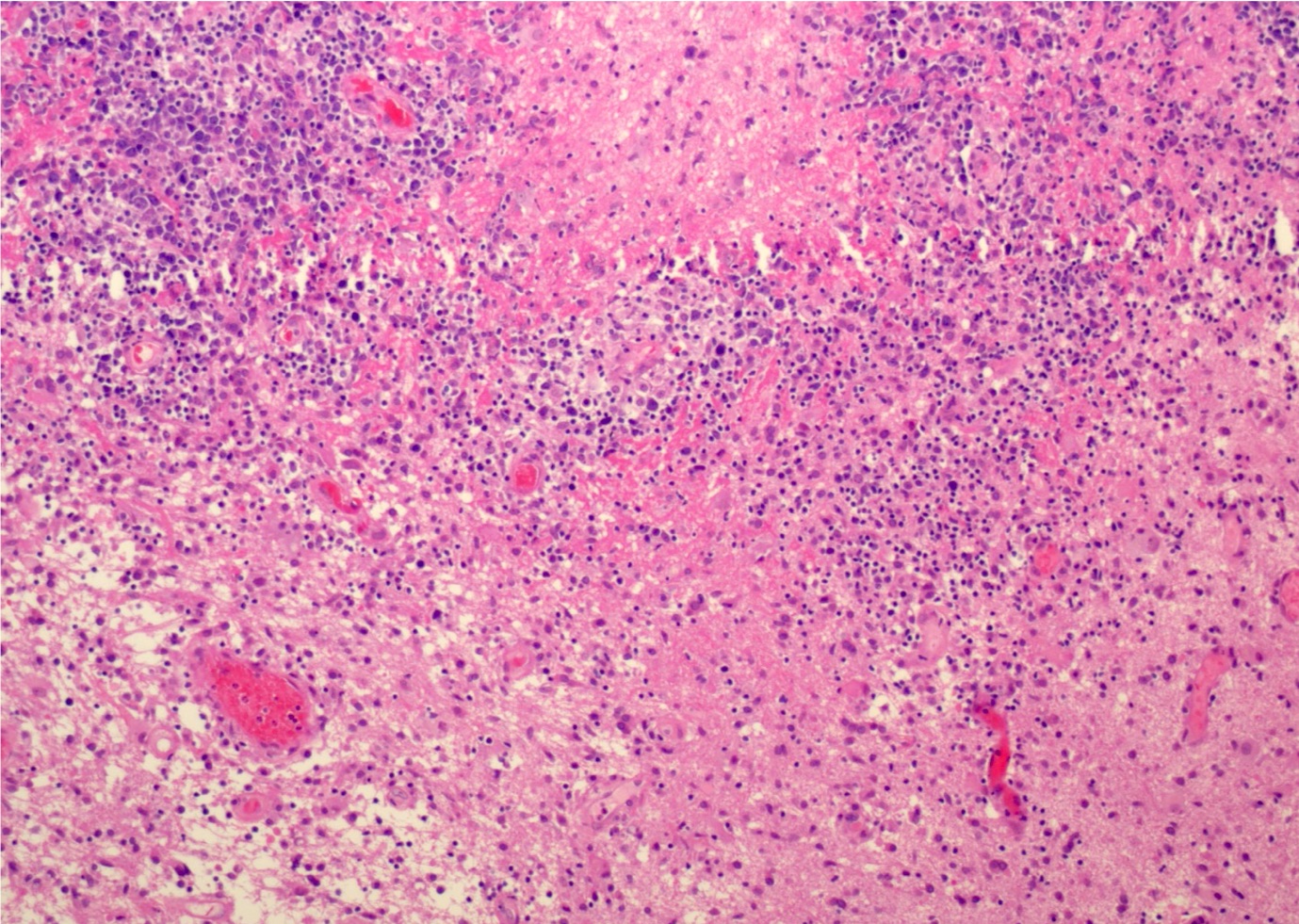
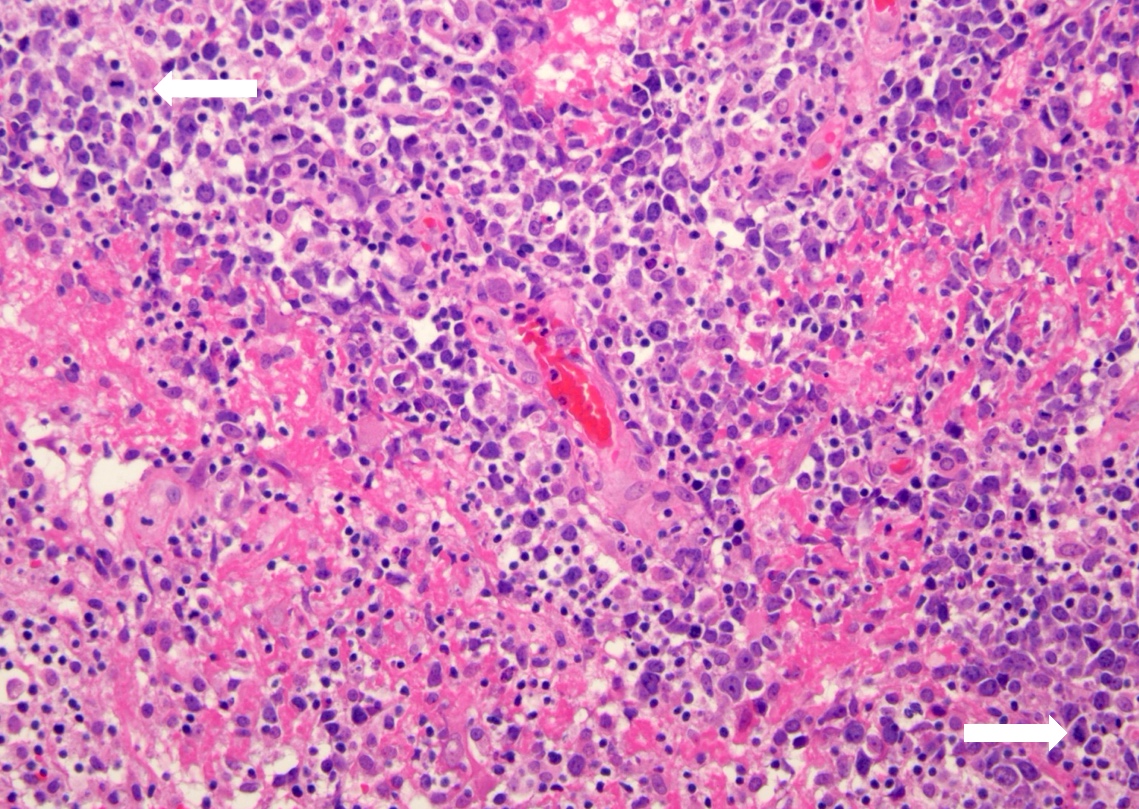
| Primary large B cell lymphoma of the central nervous system
| Primary large B cell lymphoma of immune privileged sites (new umbrella term for DLBCL arising in the CNS, vitreoretina and testis)
| Primary diffuse large B cell lymphoma of central nervous system
|
| Not included
| Primary diffuse large B cell lymphoma of testis
|
| Primary cutaneous diffuse large B cell lymphoma, leg type
| Primary cutaneous diffuse large B cell lymphoma, leg type
| Primary cutaneous diffuse large B cell lymphoma, leg type
|
| Intravascular large B cell lymphoma
| Intravascular large B cell lymphoma
| Intravascular large B cell lymphoma
|
| ALK positive large B cell lymphoma
| ALK positive large B cell lymphoma
| ALK positive large B cell lymphoma
|
| Plasmablastic lymphoma
| Plasmablastic lymphoma
| Plasmablastic lymphoma
|
| Large B cell lymphoma with IRF4 rearrangement
| Large B cell lymphoma with IRF4 rearrangement
| Large B cell lymphoma with IRF4 rearrangement
|
| Primary mediastinal large B cell lymphoma
| Primary mediastinal large B cell lymphoma
| Primary mediastinal large B cell lymphoma
|
| B cell lymphoma, unclassified with features intermediate between DLBCL and classic Hodgkin lymphoma
| Mediastinal gray zone lymphoma (cases without mediastinal involvement are classified as DLBCL, NOS)
| Mediastinal gray zone lymphoma
|
| Not included
| Fibrin associated large B cell lymphoma
| Fibrin associated large B cell lymphoma
|
| Not included
| Fluid overload associated large B cell lymphoma (previously included in primary effusion lymphoma)
| HHV8 and EBV8 negative primary effusion based lymphoma*
|
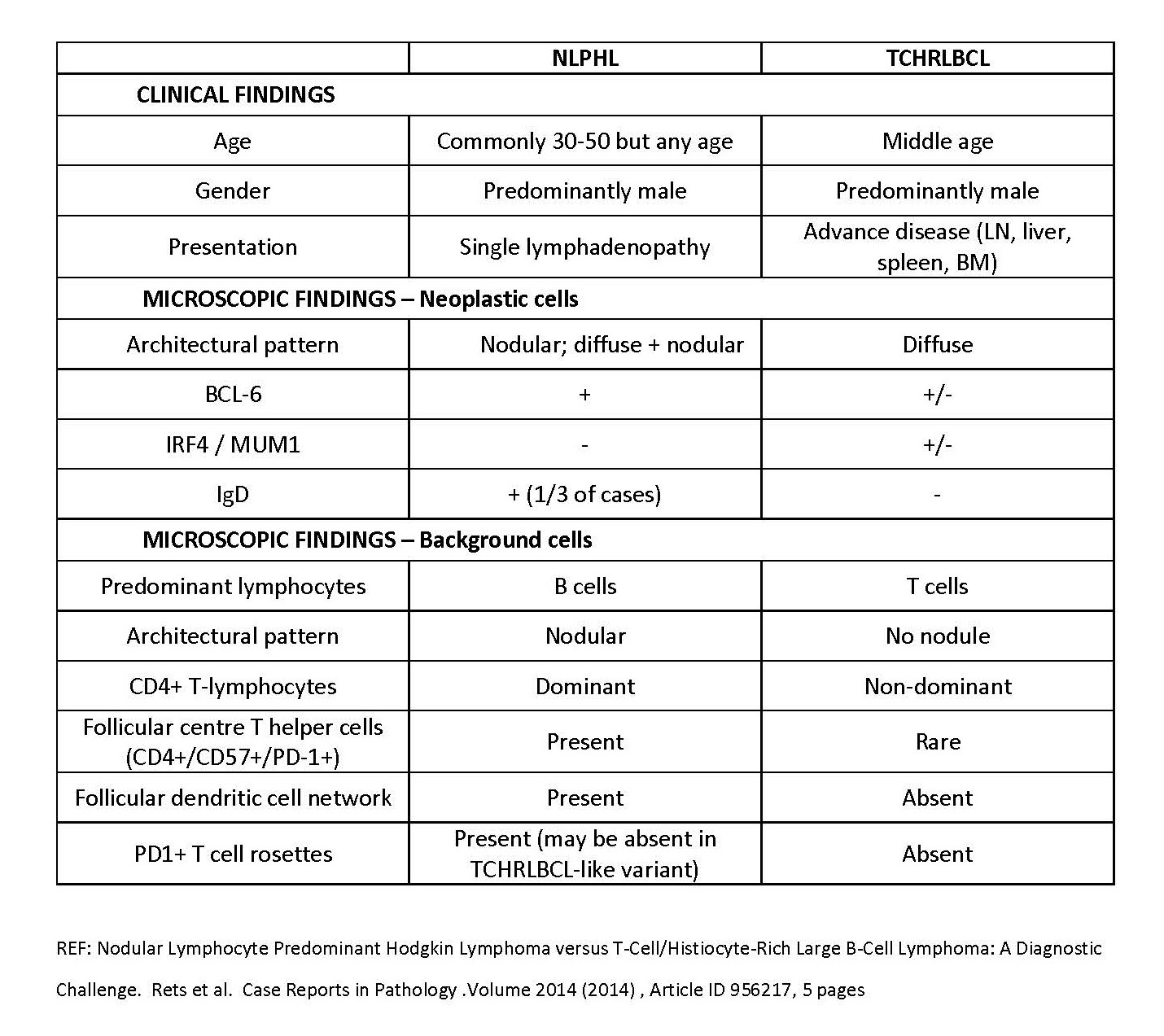
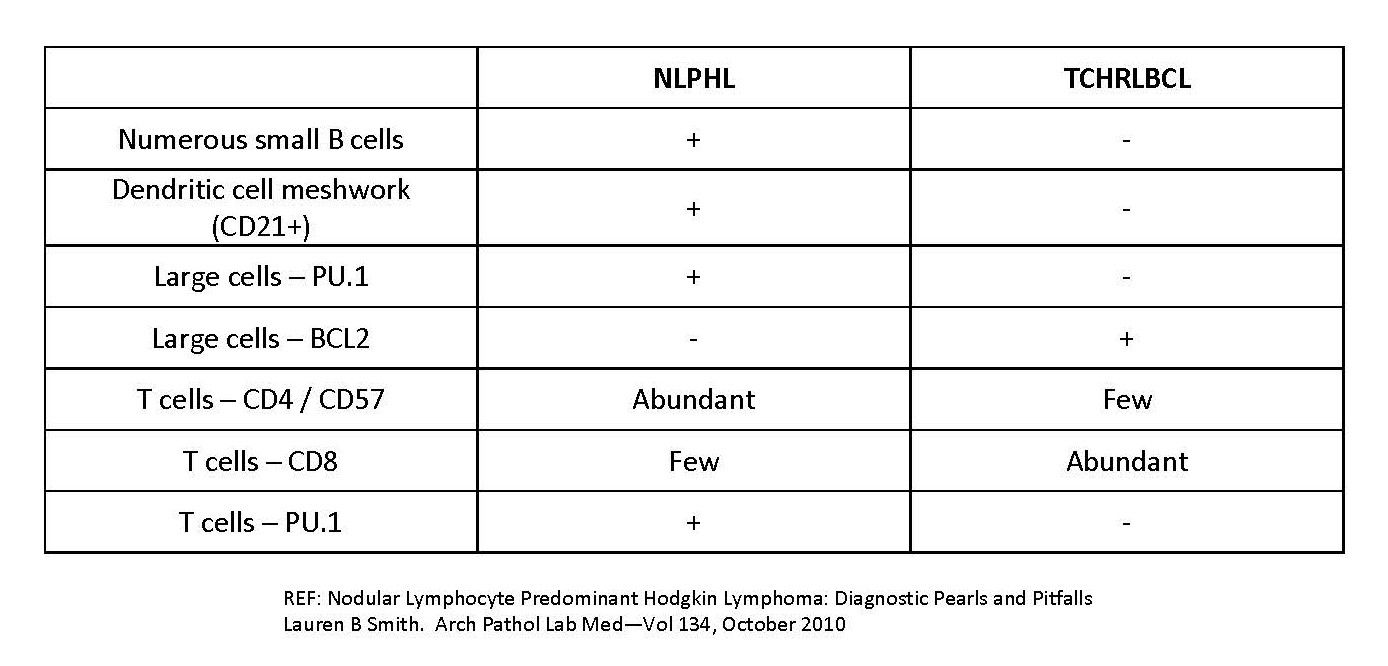
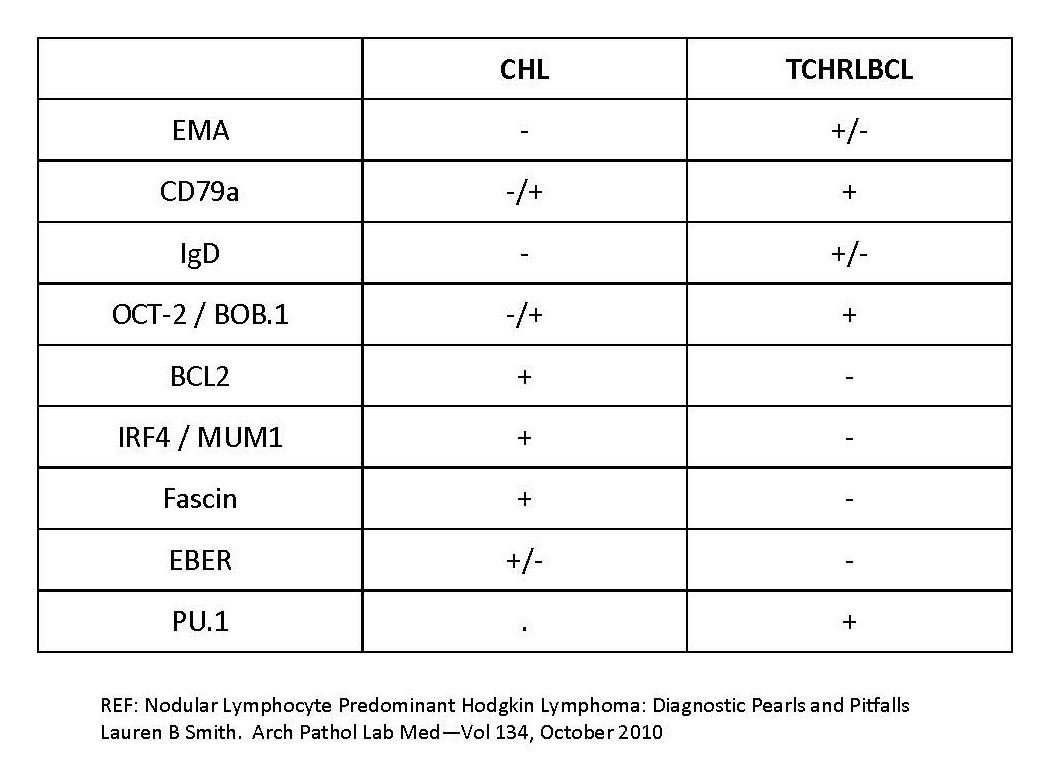
| T cell / histiocyte rich large B cell lymphoma
| T cell / histiocyte rich large B cell lymphoma
| T cell / histiocyte rich large B cell lymphoma
|
| See Hodgkin lymphomas
| See Hodgkin lymphomas
| Nodular lymphocyte predominant B cell lymphoma (renamed from nodular lymphocyte predominant Hodgkin lymphoma)
|
| Lymphomatoid granulomatosis
| Lymphomatoid granulomatosis
| Lymphomatoid granulomatosis
|
| KSHV / HHV8 associated B cell lymphoid proliferation / lymphoma
|
| Primary effusion lymphoma
| Primary effusion lymphoma
| Primary effusion lymphoma
|
| HHV8 positive diffuse large B cell lymphoma, NOS
| HHV8 positive diffuse large B cell lymphoma
| HHV8 positive diffuse large B cell lymphoma, NOS
|
| HHV8 positive germinotropic lymphoproliferative disorder
| KSHV / HHV8 positive germinotropic lymphoproliferative disorder
| HHV8 positive germinotropic lymphoproliferative disorder
|
| High grade B cell lymphomas
|
| High grade B cell lymphoma, NOS
| High grade B cell lymphoma, NOS
| High grade B cell lymphoma, NOS
|
| High grade B cell lymphoma with MYC and BCL2 or BCL6 rearrangements
| Diffuse large B cell lymphoma / high grade B cell lymphoma with MYC and BCL2 rearrangements (previous high grade B cell lymphoma with MYC and BCL6 rearrangements is designated as DLBCL, NOS)
| High grade B cell lymphoma with MYC and BCL2 rearrangements
|
| High grade B cell lymphoma with MYC and BCL6 rearrangements*
|
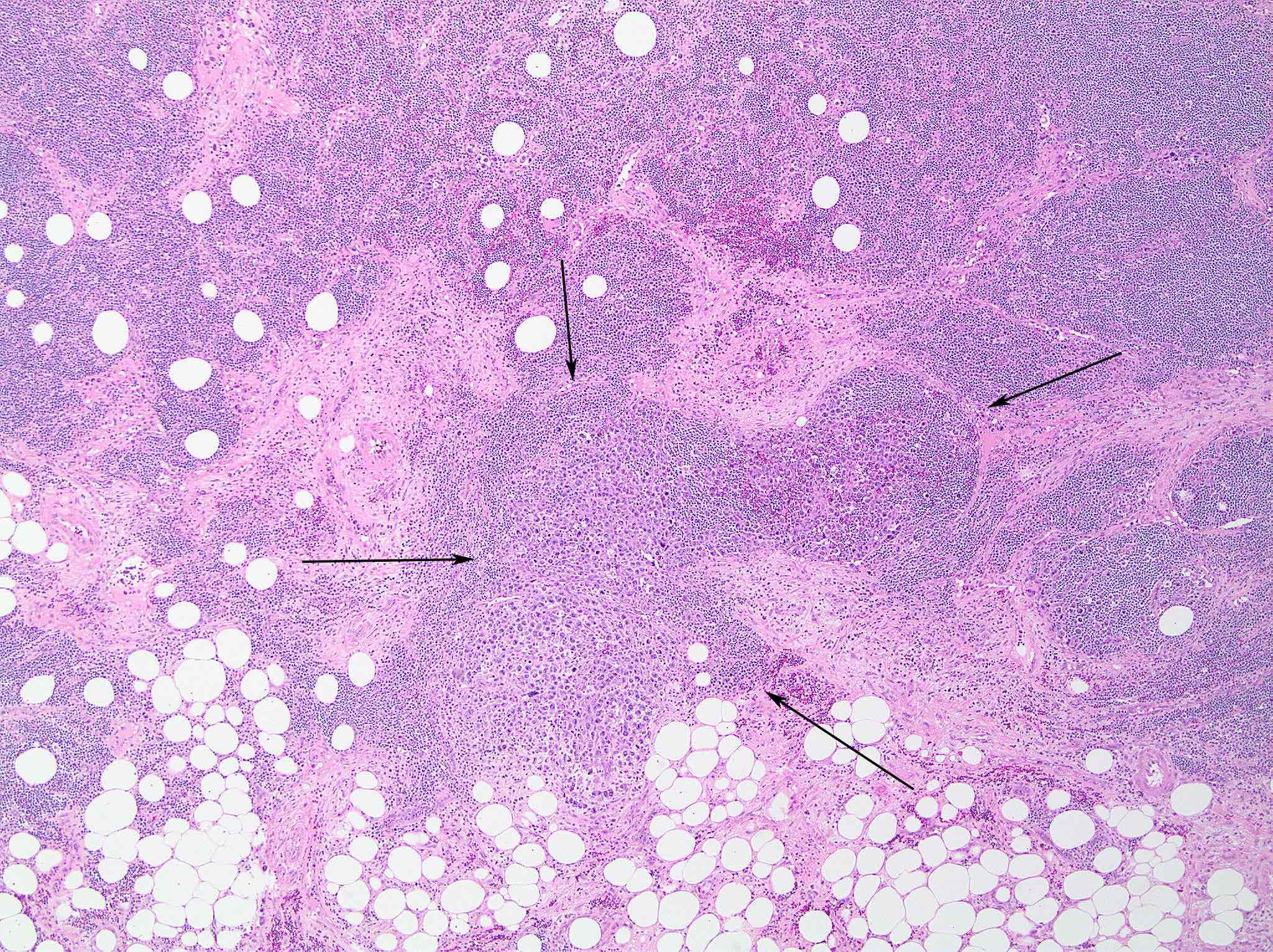
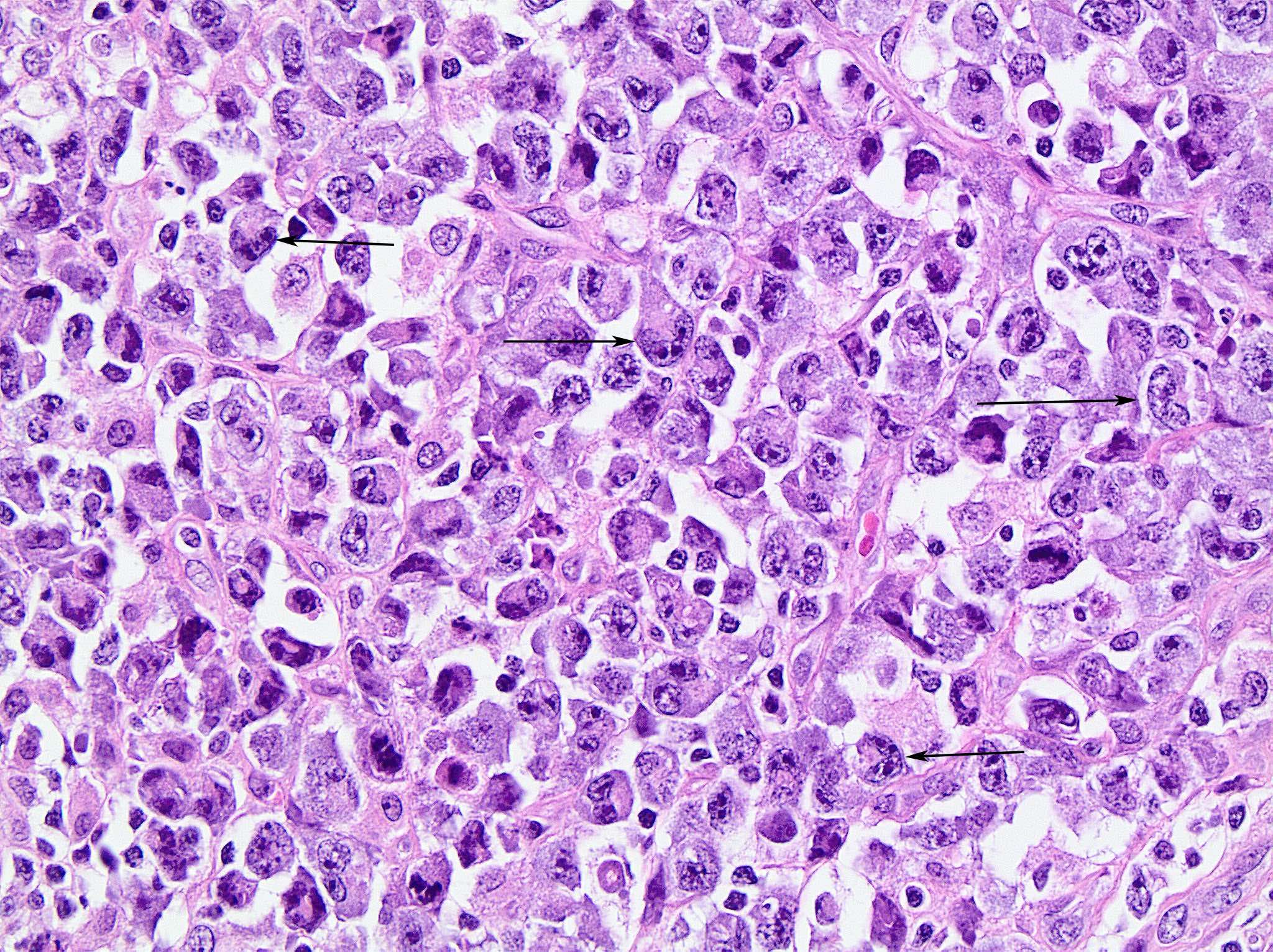
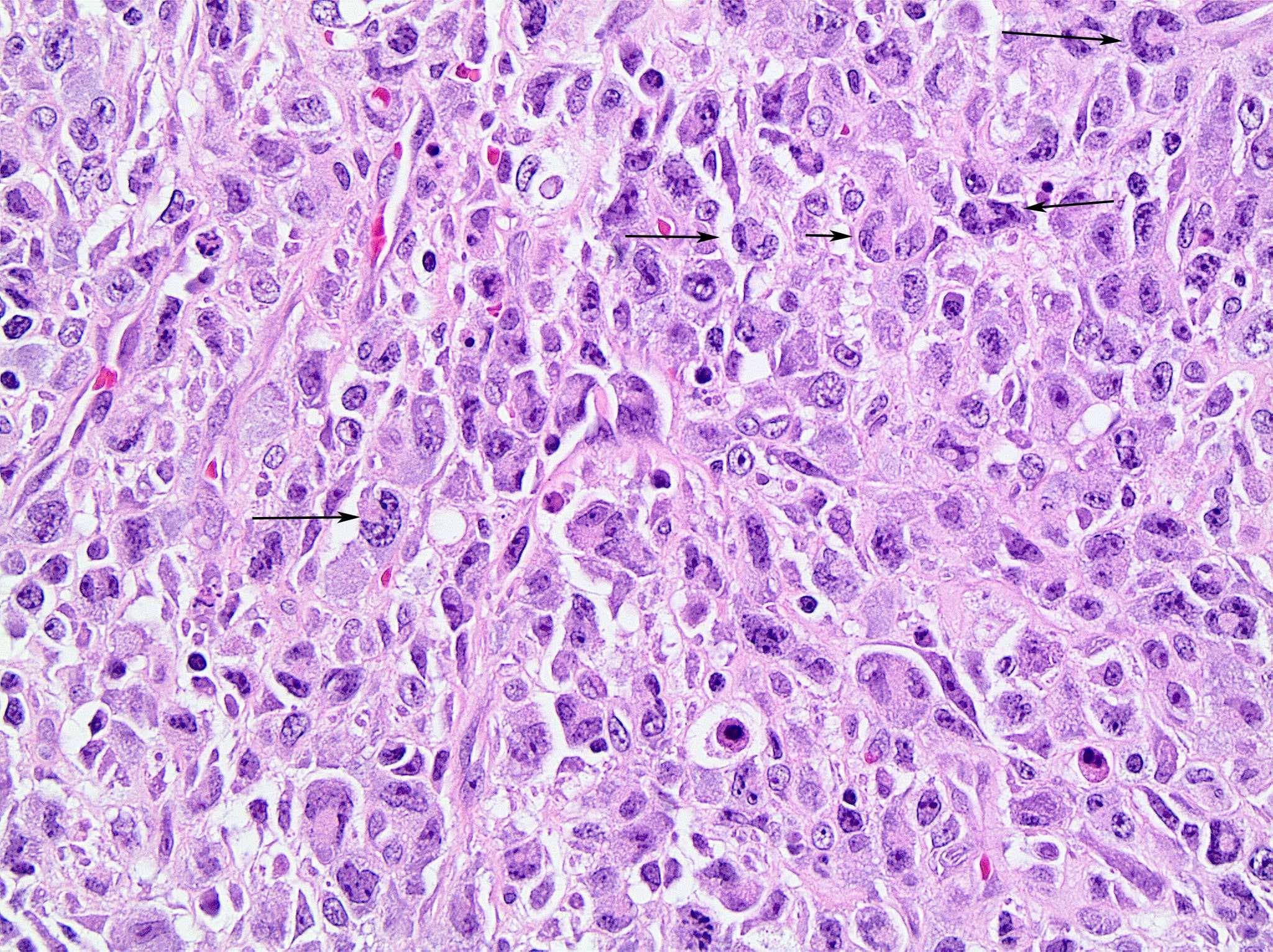
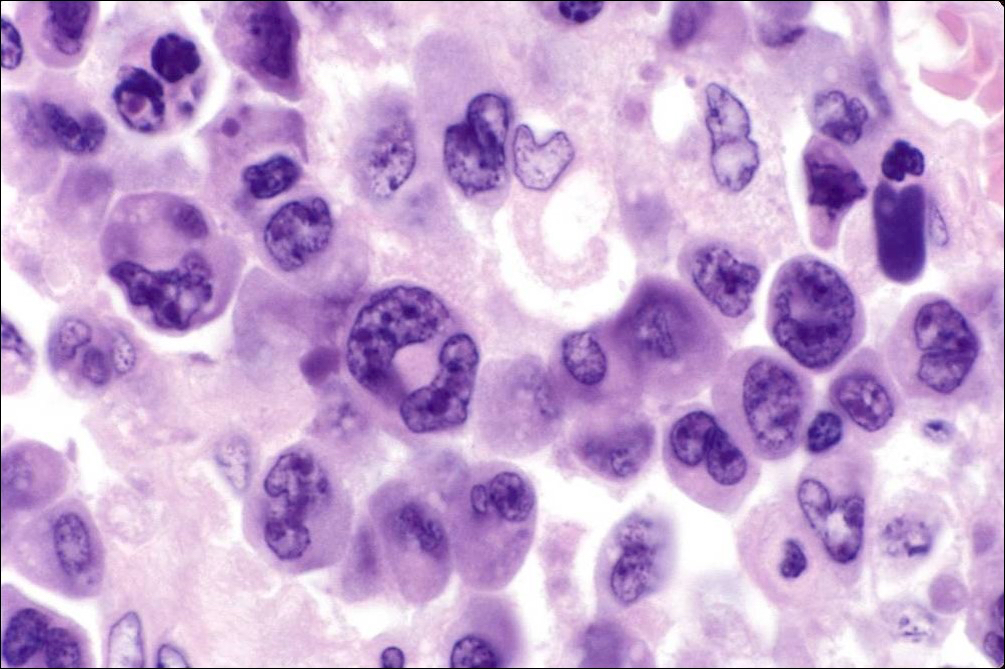
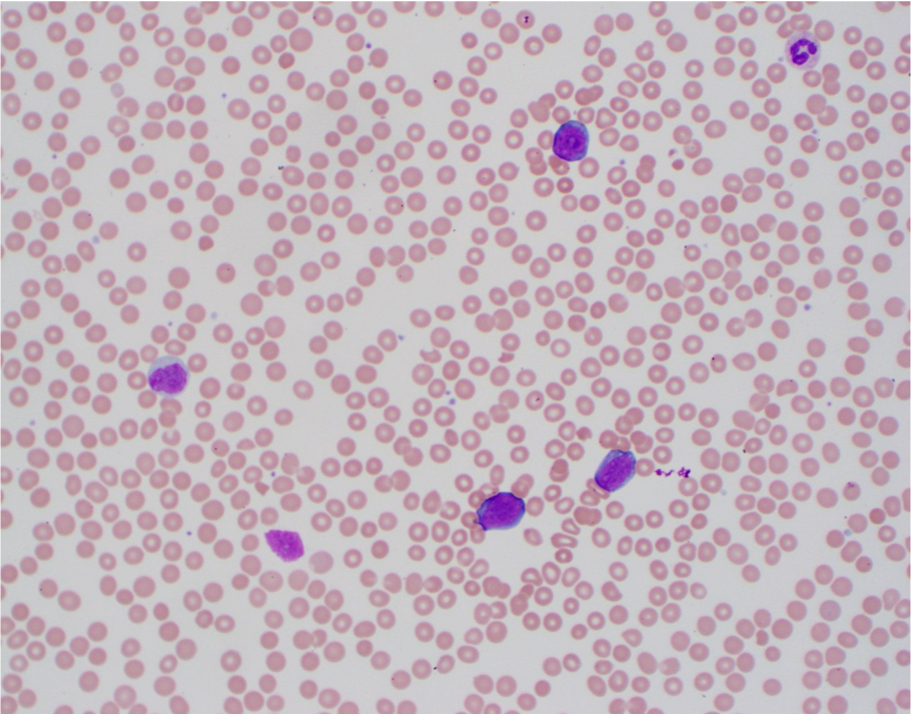
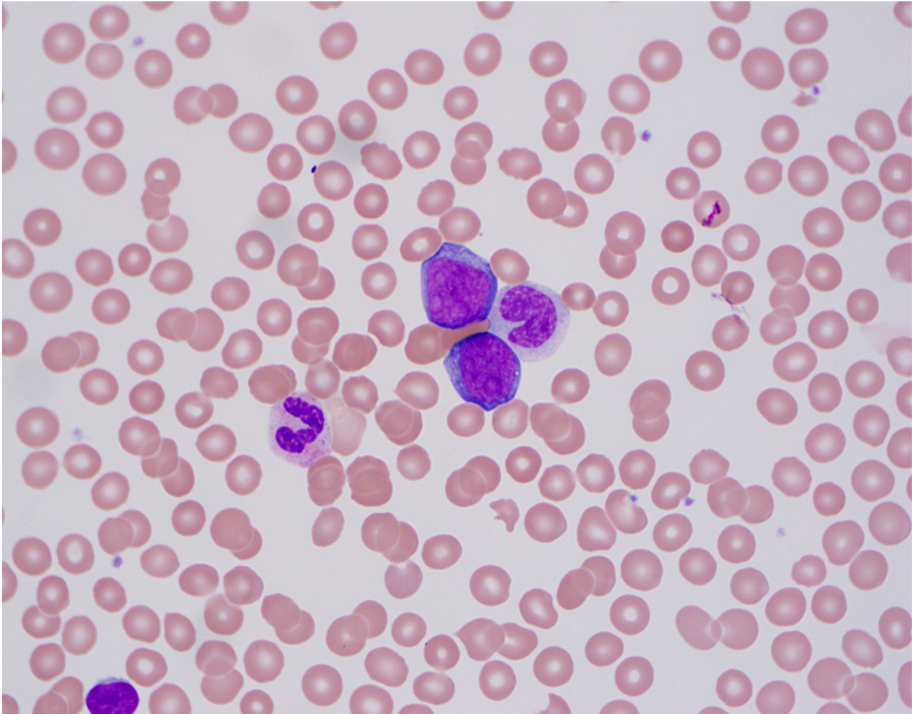
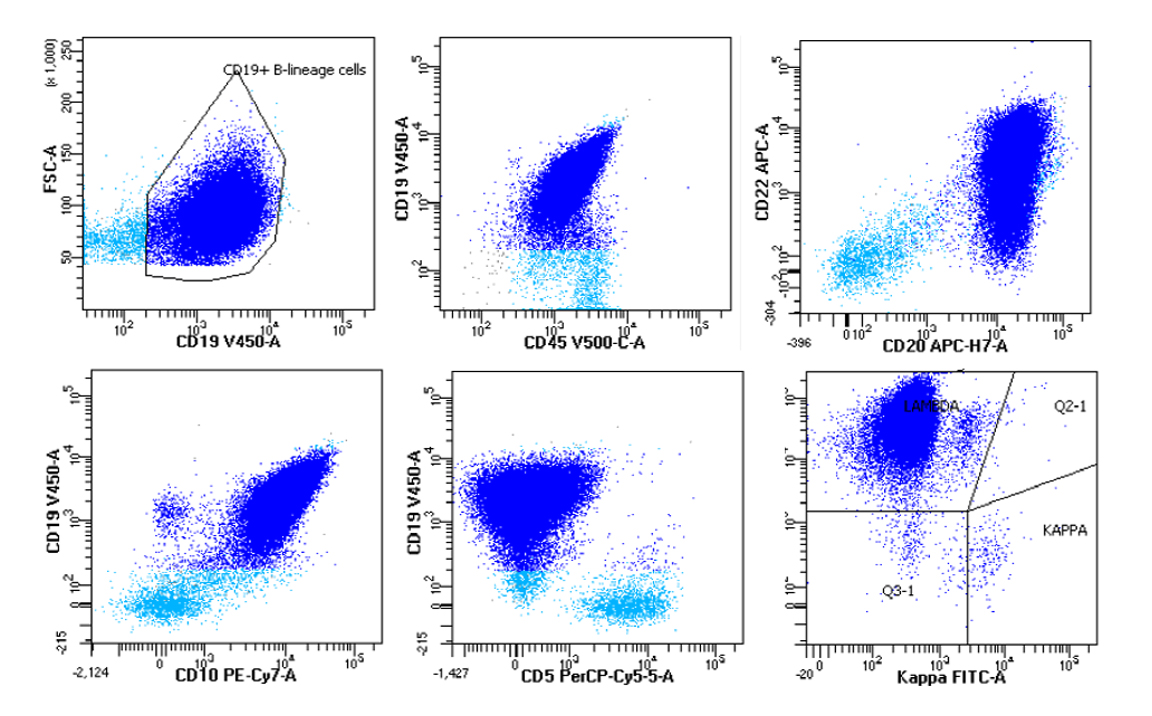
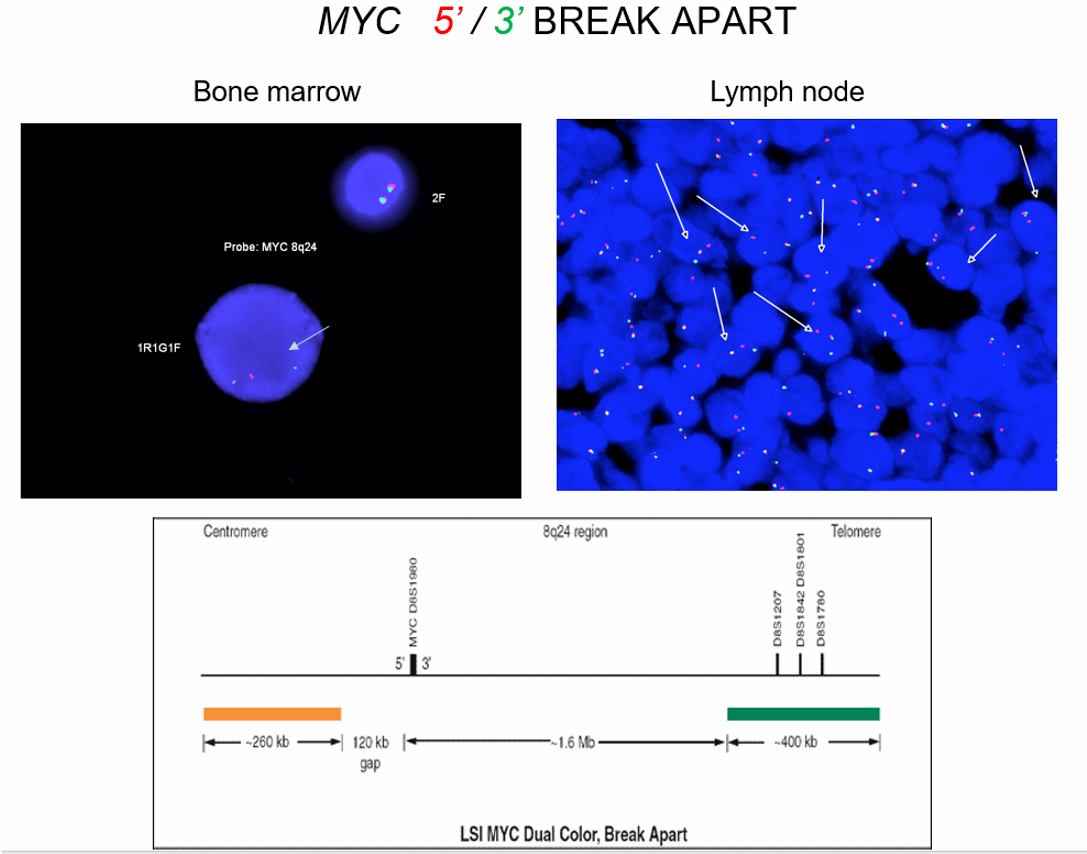
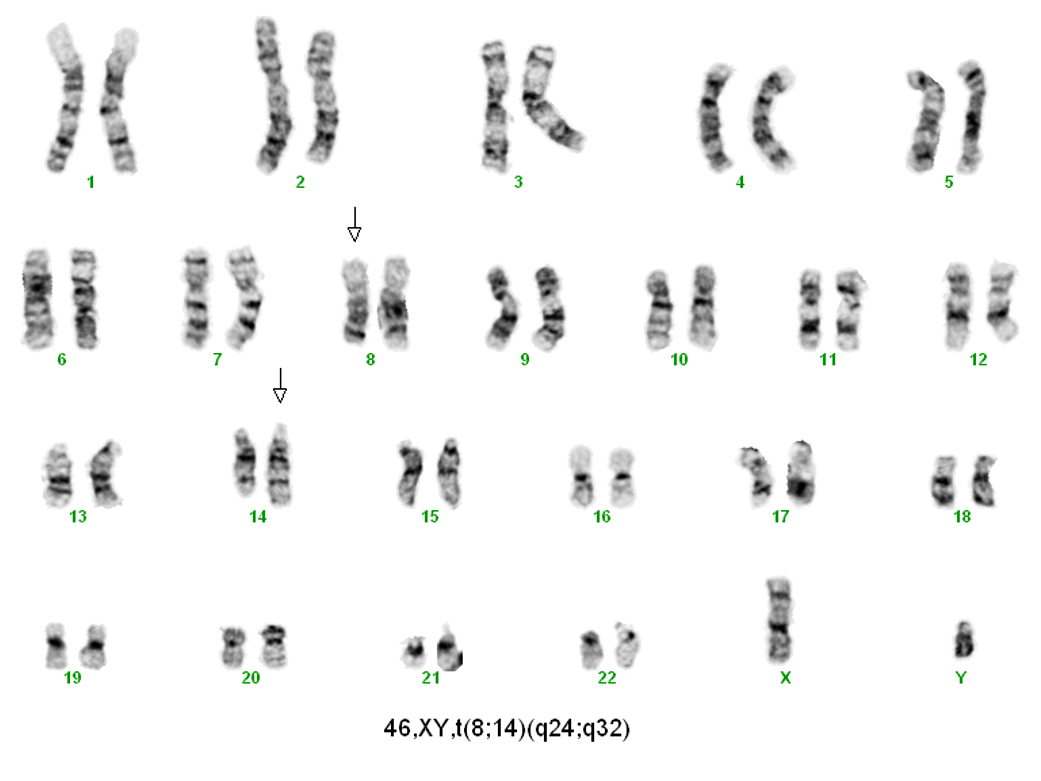
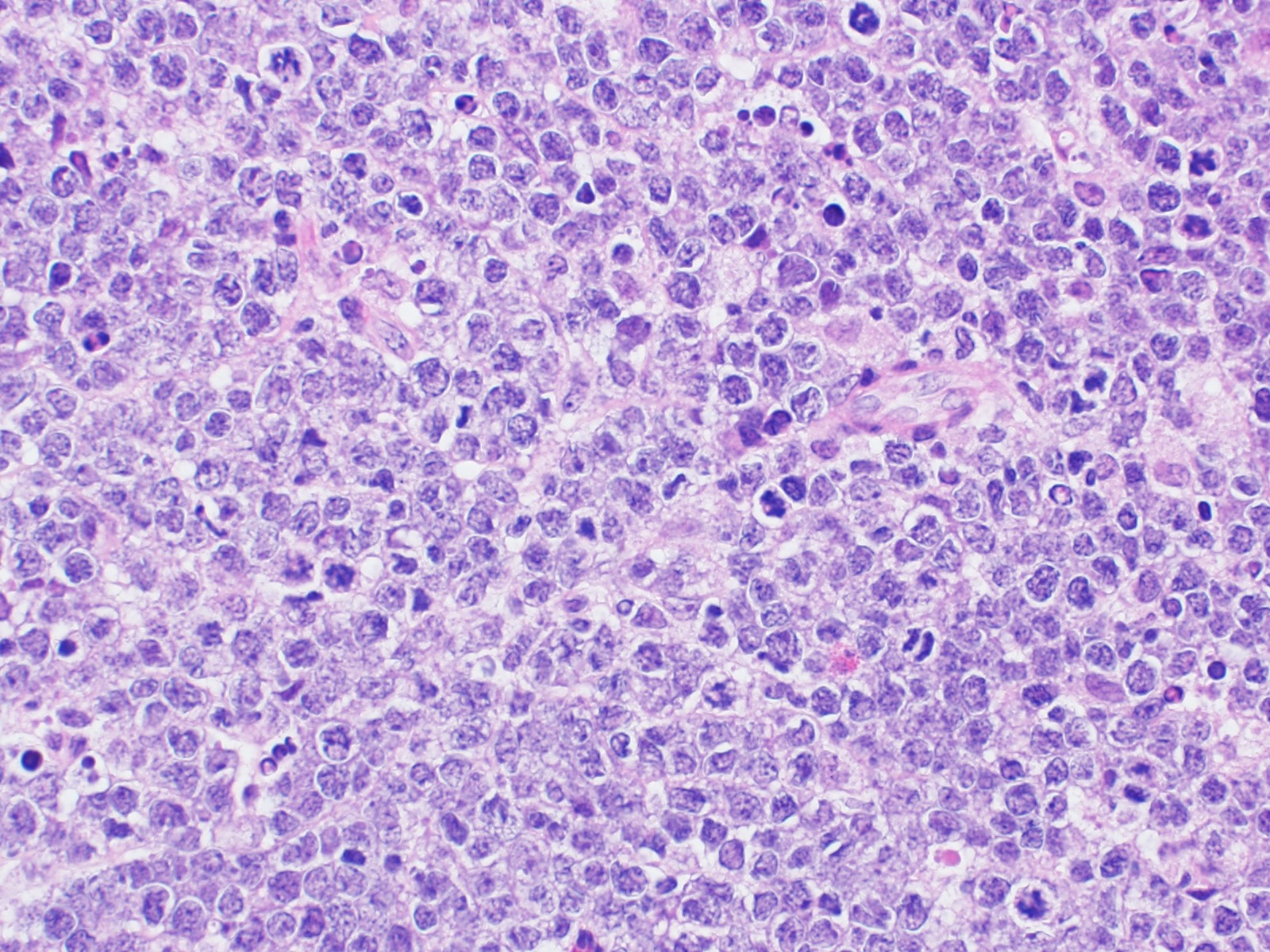
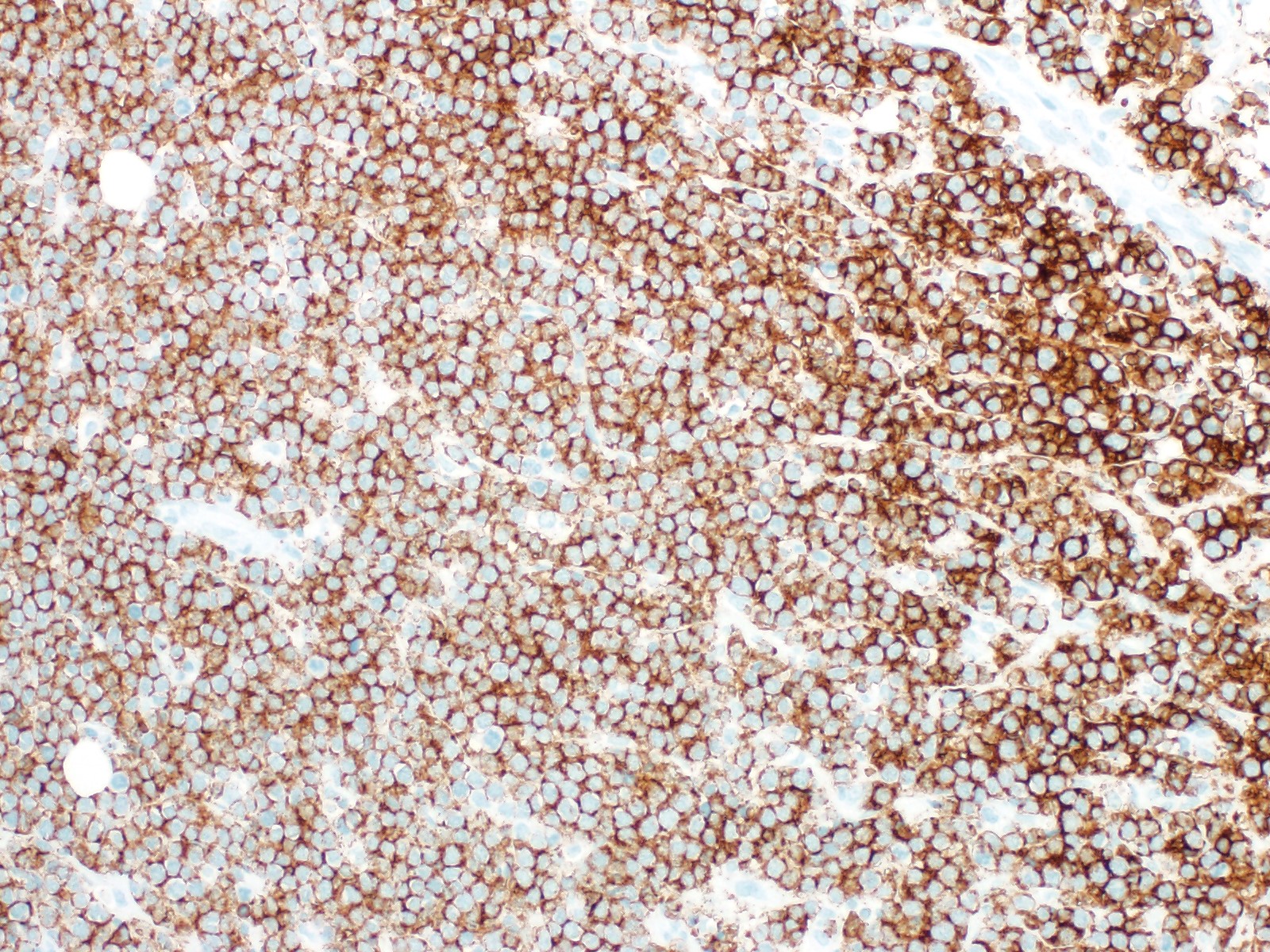
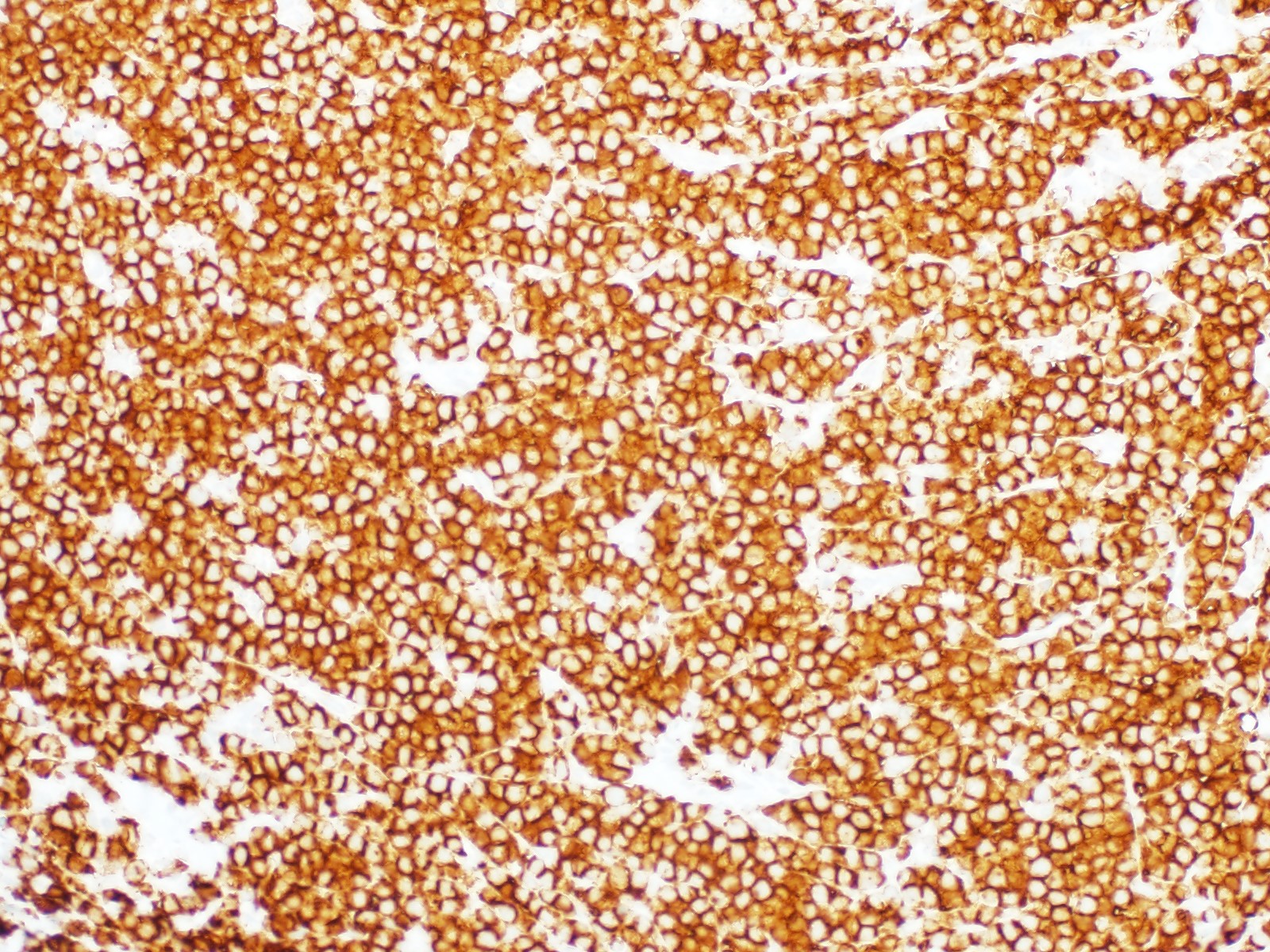
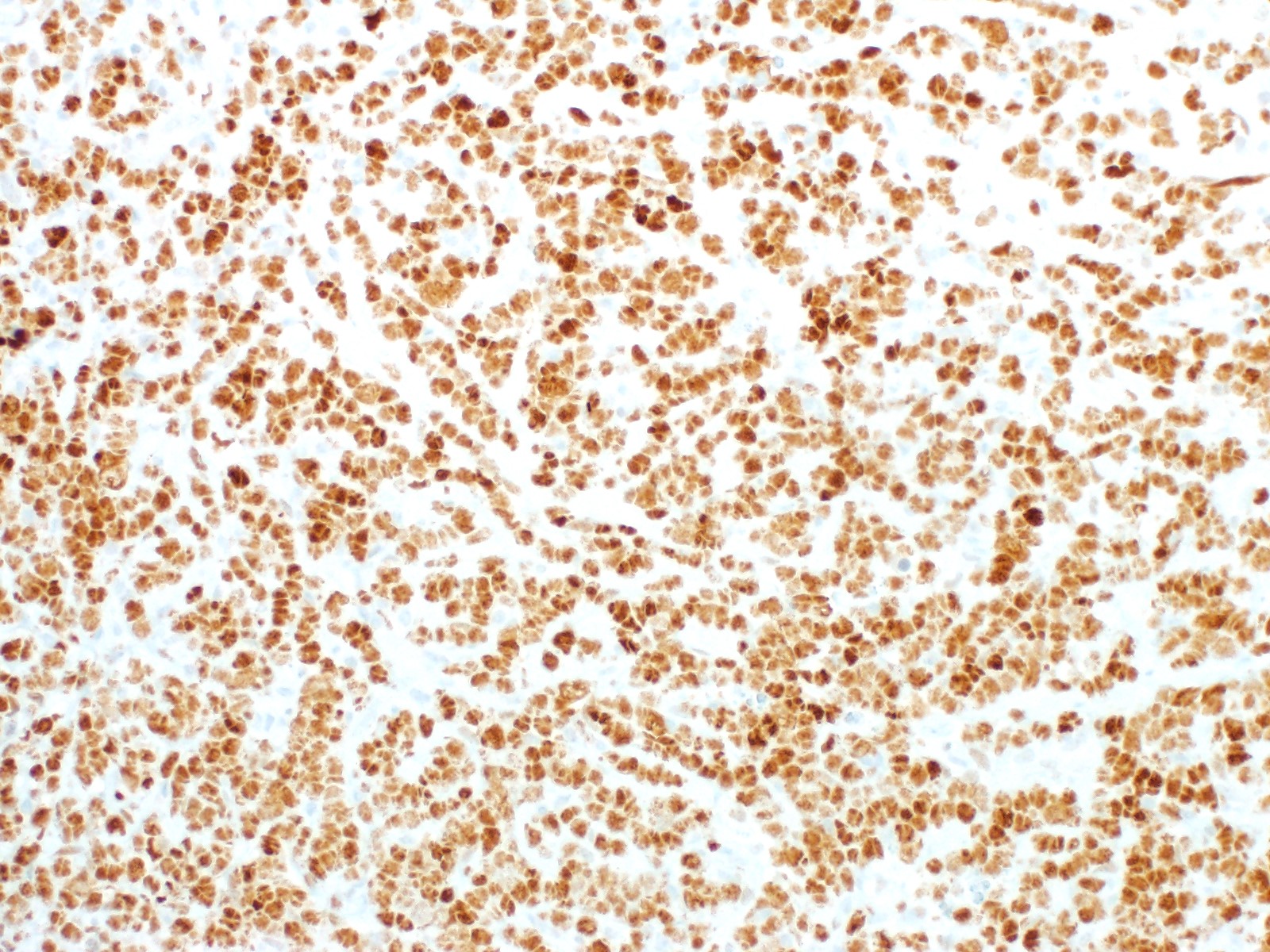
| Burkitt lymphoma
| Burkitt lymphoma (EBV status supersedes epidemiologic subtyping)
| Burkitt lymphoma
|
| Burkitt-like lymphoma with 11q aberration*
| High grade B cell lymphoma with 11q aberration
| Large B cell lymphoma with 11q aberration*
|
| Hodgkin lymphomas
|
Classic Hodgkin lymphoma - Nodular sclerosis
- Lymphocyte rich
- Mixed cellularity
- Lymphocyte depleted
| Classic Hodgkin lymphoma - Nodular sclerosis
- Lymphocyte rich
- Mixed cellularity
- Lymphocyte depleted
| Classic Hodgkin lymphoma - Nodular sclerosis
- Lymphocyte rich
- Mixed cellularity
- Lymphocyte depleted
|
| Nodular lymphocyte predominant Hodgkin lymphoma
| Nodular lymphocyte predominant Hodgkin lymphoma
| Renamed as nodular lymphocyte predominant B cell lymphoma; categorized as non-Hodgkin lymphoma
|
| Plasma cell neoplasms and entities with paraproteins
|
| Solitary plasmacytoma of bone
| Solitary plasmacytoma of bone
| Solitary plasmacytoma of bone
|
| Extraosseous plasmacytoma
| Extraosseous plasmacytoma
| Extraosseous plasmacytoma
|
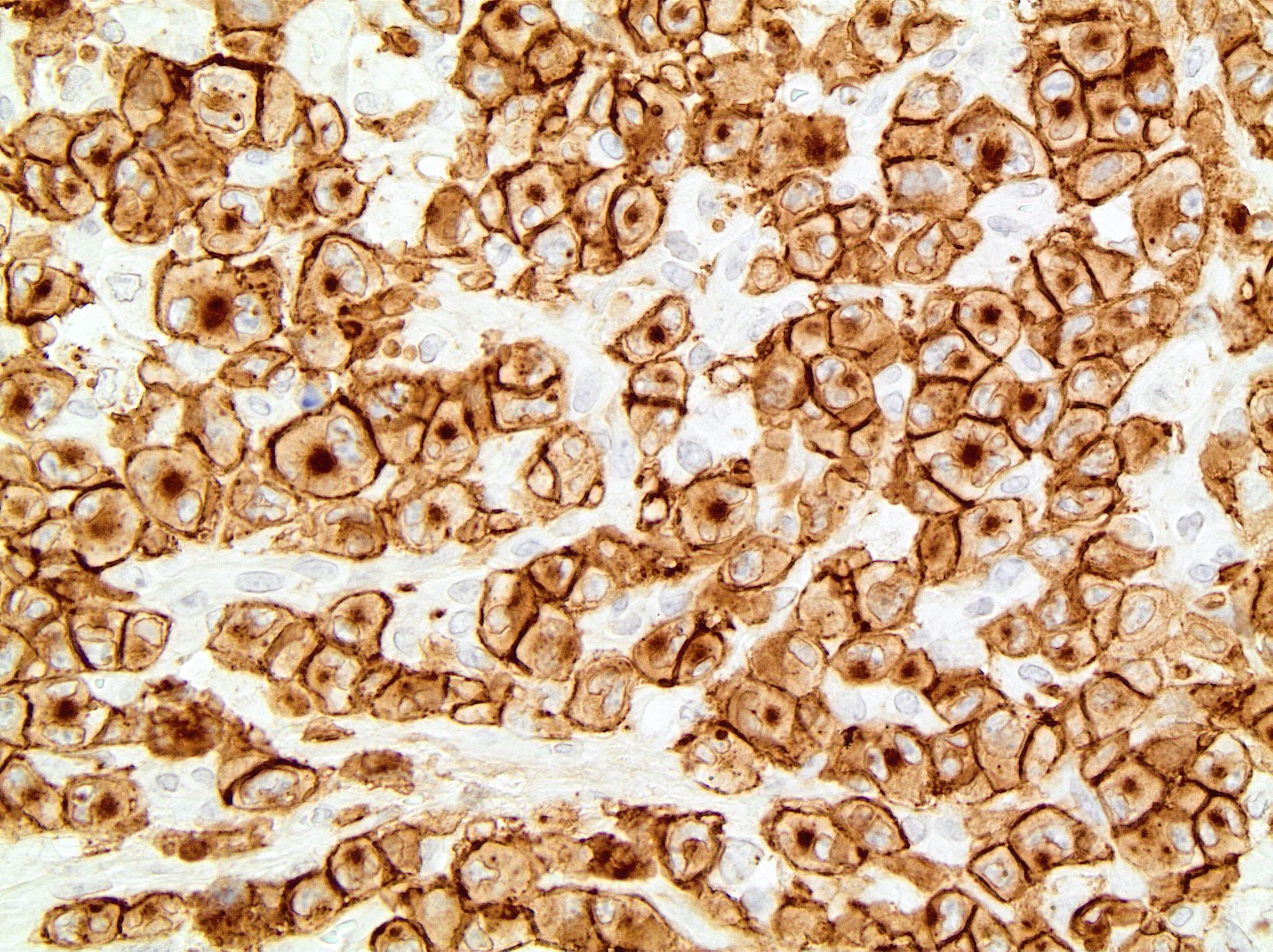
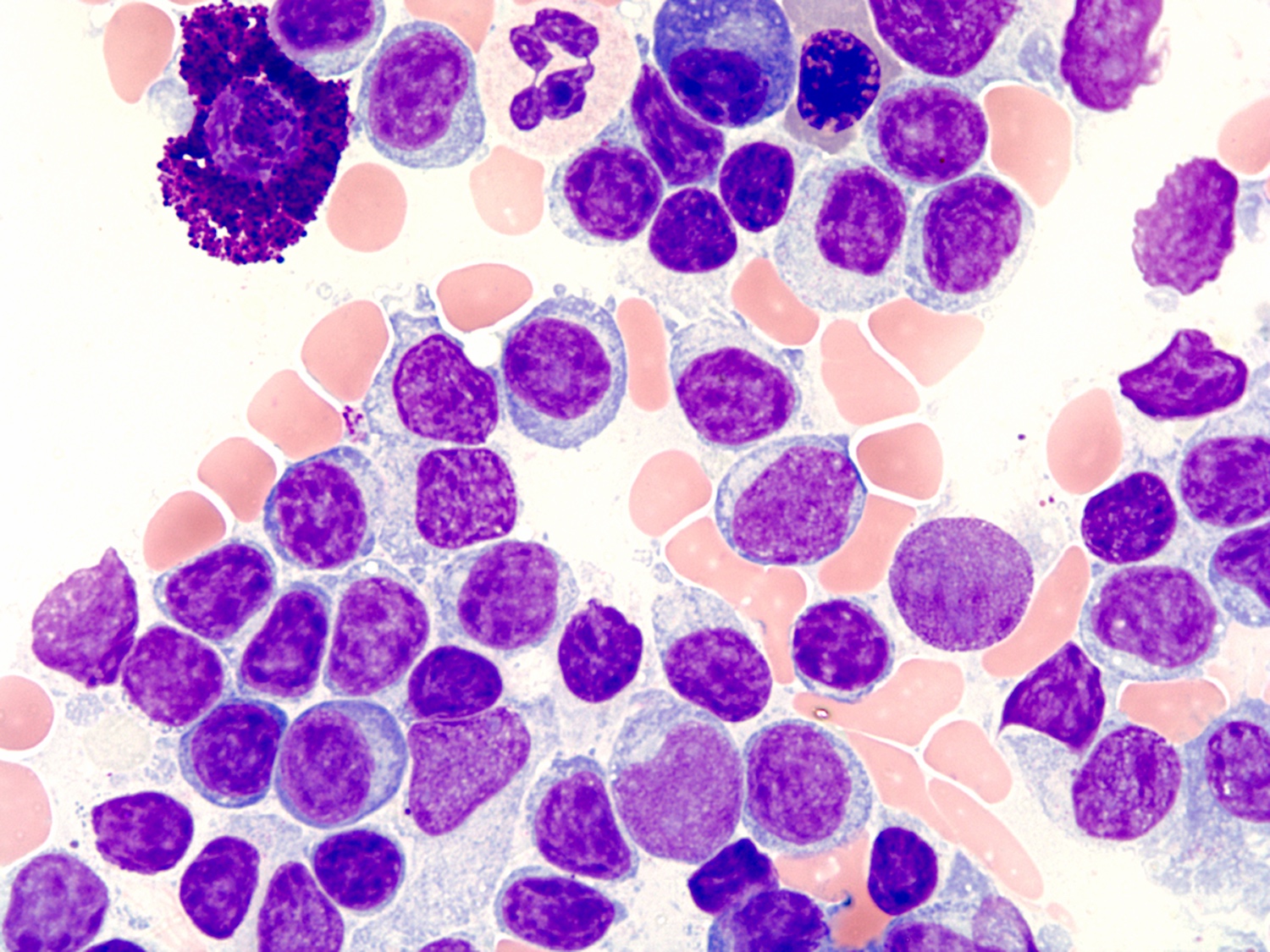
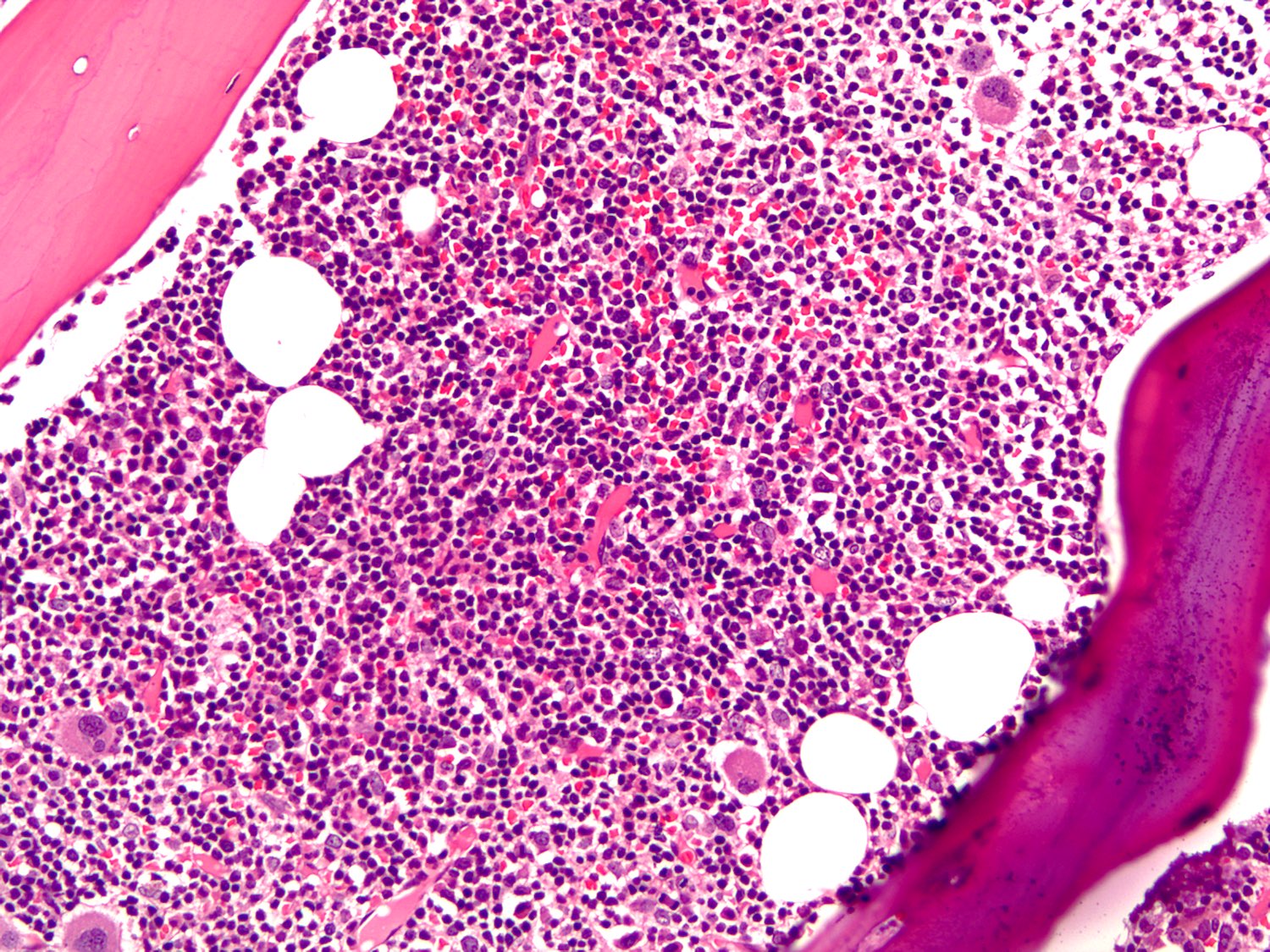
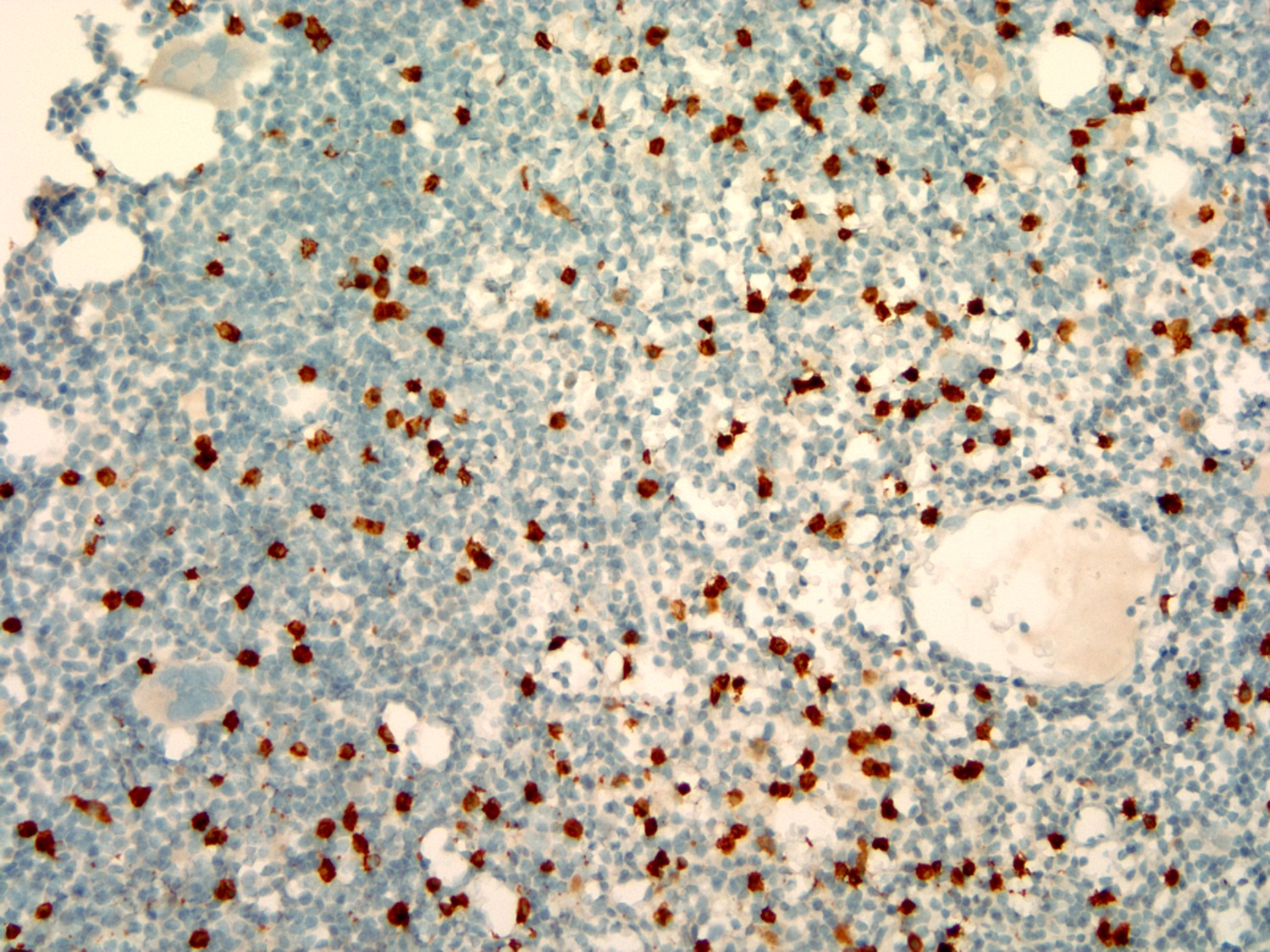
| Plasma cell myeloma
| Plasma cell myeloma
| Multiple myeloma (MM), NOS
|
MM with recurrent genetic abnormality - MM with CCND family translocation
- MM with MAF family translocation
- MM with NSD2 family translocation
- MM with hyperdiploidy
|
| Plasma cell neoplasm with associated paraneoplastic syndrome
| Plasma cell neoplasm with associated paraneoplastic syndrome - POEMS
- TEMPI
- AESOP (new syndrome)
|
|
| IgM monoclonal gammopathy of undetermined significance
| IgM monoclonal gammopathy of undetermined significance
| IgM monoclonal gammopathy of undetermined significance - IgM MGUS, plasma cell type
- IgM MGUS, NOS
|
| Non-IgM monoclonal gammopathy of undetermined significance
| Non-IgM monoclonal gammopathy of undetermined significance
| Non-IgM monoclonal gammopathy of undetermined significance
|
| Not included
| Monoclonal gammopathy of renal significance
| Not a separate entity; clinical descriptor of the underlying diagnosis (e.g., MGUS)
|
| Not included
| Cold agglutinin disease
| Primary cold agglutinin disease
|
| Primary amyloidosis
| Immunoglobulin related (AL) amyloidosis
| Immunoglobulin light chain (AL) amyloidosis
|
| Localized AL amyloidosis
|
| Light chain and heavy chain deposition disease
| Monoclonal immunoglobulin deposition disease (renamed)
| Light chain and heavy chain deposition
|
| Mu heavy chain disease
| Mu heavy chain disease
| Mu heavy chain disease
|
| Gamma heavy chain disease
| Gamma heavy chain disease
| Gamma heavy chain disease
|
| Alpha heavy chain disease
| Alpha heavy chain disease
| Alpha heavy chain disease
|
| Lymphoid proliferations / lymphomas with immune deficiency or dysregulation
|
| Nondestructive PTLD
| Hyperplasias arising in immune deficiency / dysregulation
| Plasmacytic hyperplasia PTLD
|
| Florid follicular hyperplasia PTLD
|
| Infectious mononucleosis PTLD
|
| Polymorphic PTLD
| Polymorphic lymphoproliferative disorders arising in immune deficiency / dysregulation (new term that includes various etiologies)
| Polymorphic PTLD
|
| Other iatrogenic immunodeficiency associated lymphoproliferative disorders | Other iatrogenic immunodeficiency associated lymphoproliferative disorders
|
| Monomorphic PTLD
| Lymphomas arising in immune deficiency / dysregulation (new umbrella term that includes monomorphic PTLD, lymphomas associated with HIV infection, etc.)
| Monomorphic PTLD
|
| Classic Hodgkin lymphoma PTLD
| Classic Hodgkin lymphoma PTLD
|
| Lymphomas associated with HIV infection
|
|
| Lymphoproliferative diseases associated with primary immune disorders
| Inborn error of immunity associated lymphoid proliferations and lymphomas
|
|